Upgrade to Pro
— share decks privately, control downloads, hide ads and more …
Speaker Deck
Features
Speaker Deck
PRO
Sign in
Sign up for free
Search
Search
ゲノム解析における射影: 特徴選択ツールとしてのテンソル分解と主成分分析を合理化する理論的根拠
Search
Y-h. Taguchi
June 22, 2025
Science
0
62
ゲノム解析における射影: 特徴選択ツールとしてのテンソル分解と主成分分析を合理化する理論的根拠
Presentation at SOGBIO82
https://www.ipsj.or.jp/kenkyukai/event/mps153bio82.html
Y-h. Taguchi
June 22, 2025
Tweet
Share
More Decks by Y-h. Taguchi
See All by Y-h. Taguchi
presen_同仁倶楽部.pdf
tagtag
0
24
知能とはなにか -ヒトとAIのあいだ-
tagtag
1
52
生成AIの現状と展望
tagtag
0
71
主成分分析に基づく教師なし特徴抽出法を用いたコラーゲン-グリコサミノグリカンメッシュの遺伝子発現への影響
tagtag
0
160
中央大学AI・データサイエンスセンター 2025年第6回イブニングセミナー 『知能とはなにか ヒトとAIのあいだ』
tagtag
0
100
AI(人工知能)の過去・現在・未来 —AIは人間を超えるのか—
tagtag
0
66
知能とはなにかーヒトとAIのあいだー
tagtag
0
160
タンパク質間相互作⽤を利⽤した⼈⼯知能による新しい薬剤遺伝⼦-疾患相互作⽤の同定
tagtag
0
130
PPIのみを用いたAIによる薬剤–遺伝子–疾患 相互作用の同定
tagtag
0
120
Other Decks in Science
See All in Science
白金鉱業Vol.21【初学者向け発表枠】身近な例から学ぶ数理最適化の基礎 / Learning the Basics of Mathematical Optimization Through Everyday Examples
brainpadpr
1
490
MCMCのR-hatは分散分析である
moricup
0
540
データベース06: SQL (3/3) 副問い合わせ
trycycle
PRO
1
710
高校生就活へのDA導入の提案
shunyanoda
0
6.1k
Hakonwa-Quaternion
hiranabe
1
160
防災デジタル分野での官民共創の取り組み (1)防災DX官民共創をどう進めるか
ditccsugii
0
440
NDCG is NOT All I Need
statditto
2
2.6k
DMMにおけるABテスト検証設計の工夫
xc6da
1
1.4k
機械学習 - DBSCAN
trycycle
PRO
0
1.4k
データベース03: 関係データモデル
trycycle
PRO
1
320
力学系から見た現代的な機械学習
hanbao
3
3.7k
安心・効率的な医療現場の実現へ ~オンプレAI & ノーコードワークフローで進める業務改革~
siyoo
0
430
Featured
See All Featured
The Impact of AI in SEO - AI Overviews June 2024 Edition
aleyda
5
680
The Pragmatic Product Professional
lauravandoore
37
7.1k
Self-Hosted WebAssembly Runtime for Runtime-Neutral Checkpoint/Restore in Edge–Cloud Continuum
chikuwait
0
240
A Tale of Four Properties
chriscoyier
162
23k
Leveraging LLMs for student feedback in introductory data science courses - posit::conf(2025)
minecr
0
93
We Have a Design System, Now What?
morganepeng
54
7.9k
Being A Developer After 40
akosma
91
590k
Applied NLP in the Age of Generative AI
inesmontani
PRO
3
2k
How to Build an AI Search Optimization Roadmap - Criteria and Steps to Take #SEOIRL
aleyda
1
1.8k
Accessibility Awareness
sabderemane
0
24
Why Mistakes Are the Best Teachers: Turning Failure into a Pathway for Growth
auna
0
29
The Curse of the Amulet
leimatthew05
0
4.8k
Transcript
1/23: はじめに - ゲノムビッグデータの挑戦 現代の生命科学は、膨大なゲノムデータを扱います。 このデータから、生命現象や疾患の鍵となる少数の重要な遺伝子を見つけ出すことが、極めて重要です。 「特徴選択」における大きな壁 遺伝子の数 (N) がサンプル数
(M) を圧倒的に上回る ($N \gg M$) 例: 20,000遺伝子 vs 100サンプル これは「次元の呪い」とも呼ばれ、統計的な解析を著しく困難にします。 本日は、この課題に対する強力なアプローチとその理論的背景についてお話しします。
None
2/23: 従来手法とその限界 これまで、発現量が変動する遺伝子(DEG)の同定には、統計検定(t検定など)が広く用いられてきました。 従来手法の根本的な問題点 p値のサンプルサイズ依存性 生物学的な差が小さくても、サンプル数(M)を増やせば、p値はいくらでも小さくなり「統計的に有 意」という結果が出てしまいます。 これは、結果の信頼性を損なう大きな要因です。 この問題を補うため、Fold Change(発現変動幅)などの基準が併用されますが、場当たり的な解決策
に過ぎません。 より本質的で、頑健な手法が求められています。
3/23: 新しい潮流:教師なし特徴抽出 近年、統計検定に代わるアプローチとして、教師なし特徴抽出が注目を集めています。 主成分分析 (PCA) や テンソル分解 (TD) を利用します。 これらの手法は、サンプルに付随する「がん/正常」のようなラベル情報を一切
使わずに、データ自身の構造から本質的な情報を抽出します。 多くの研究で、従来法を上回る優れた性能を示すことが報告されています。 データ自身の声に耳を傾けるアプローチです。
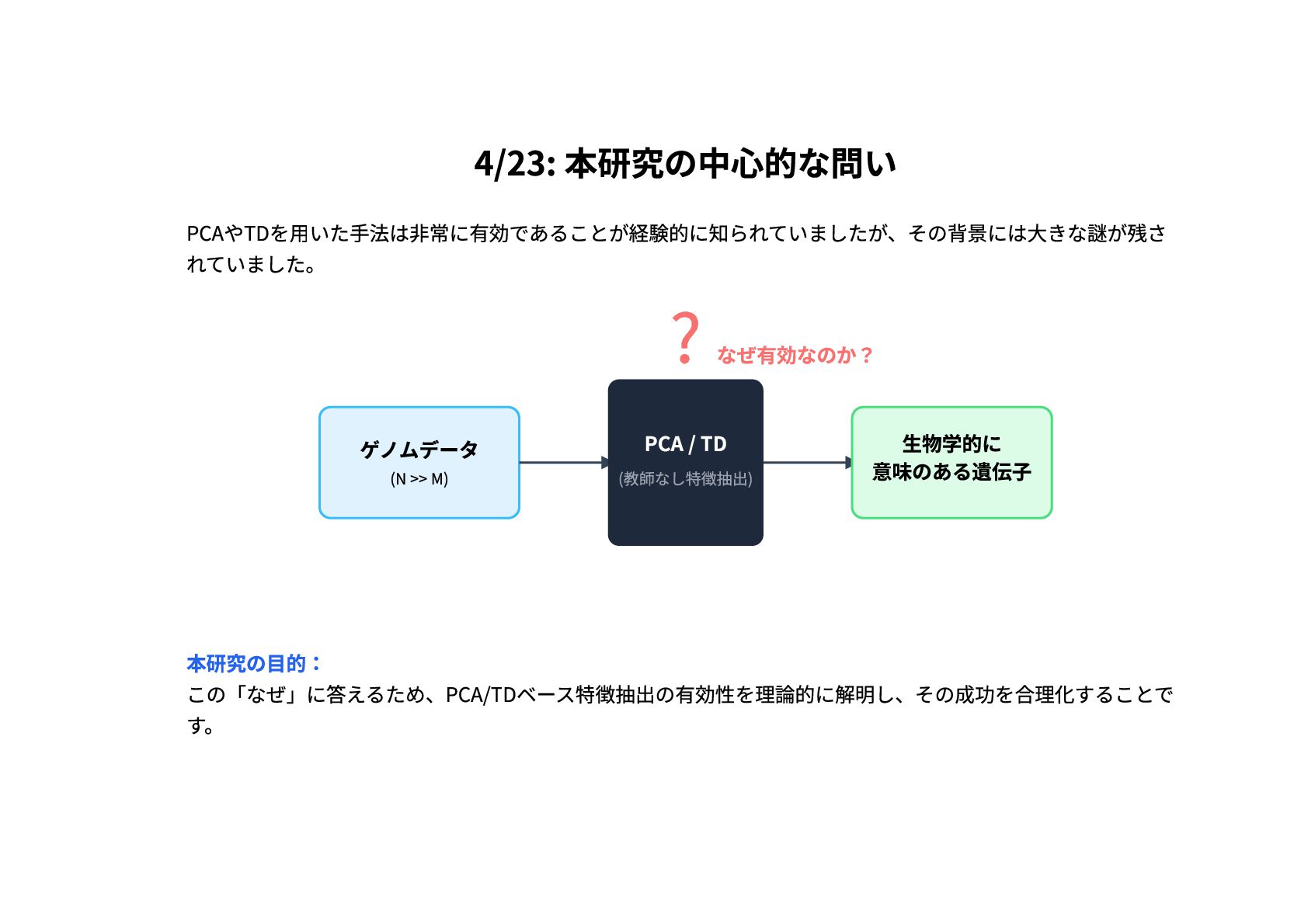
4/23: 本研究の中心的な問い PCAやTDを用いた手法は非常に有効であることが経験的に知られていましたが、その背景には大きな謎が残さ れていました。 ゲノムデータ (N >> M) PCA /
TD (教師なし特徴抽出) ? なぜ有効なのか? 生物学的に 意味のある遺伝子 本研究の目的: この「なぜ」に答えるため、PCA/TDベース特徴抽出の有効性を理論的に解明し、その成功を合理化することで す。
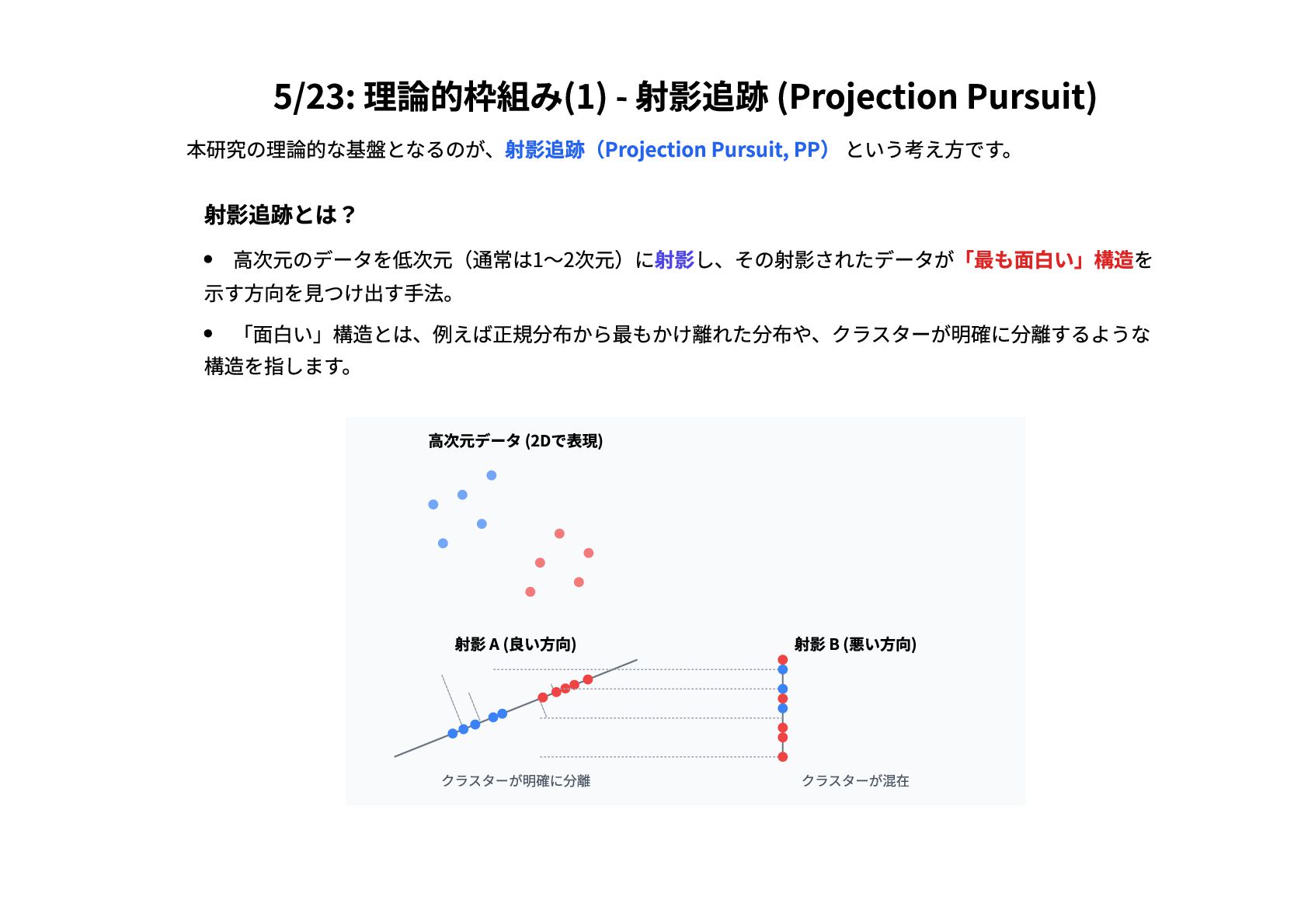
5/23: 理論的枠組み(1) - 射影追跡 (Projection Pursuit) 本研究の理論的な基盤となるのが、射影追跡(Projection Pursuit, PP) という考え方です。
射影追跡とは? 高次元のデータを低次元(通常は1〜2次元)に射影し、その射影されたデータが「最も面白い」構造を 示す方向を見つけ出す手法。 「面白い」構造とは、例えば正規分布から最もかけ離れた分布や、クラスターが明確に分離するような 構造を指します。 高次元データ (2Dで表現) 射影 A (良い方向) クラスターが明確に分離 射影 B (悪い方向) クラスターが混在
6/23: 理論的枠組み(2) - PCAとK-meansクラスタリング 射影追跡の考え方を理解する上で、PCAとK-meansクラスタリングの関係が鍵となります。 K-meansクラスタリング データをK個のグループに分類し、各グループの 中心(セントロイド)を計算する手法。 主成分分析 (PCA)
データの分散が最大になる方向(主成分)を見 つける手法。 重要な理論的背景 (Ding & He, 2004) PCAによって得られる主成分ベクトルが張る空間は、K-meansで最適化されたクラスターのセントロイド が張る空間と一致することが知られています。
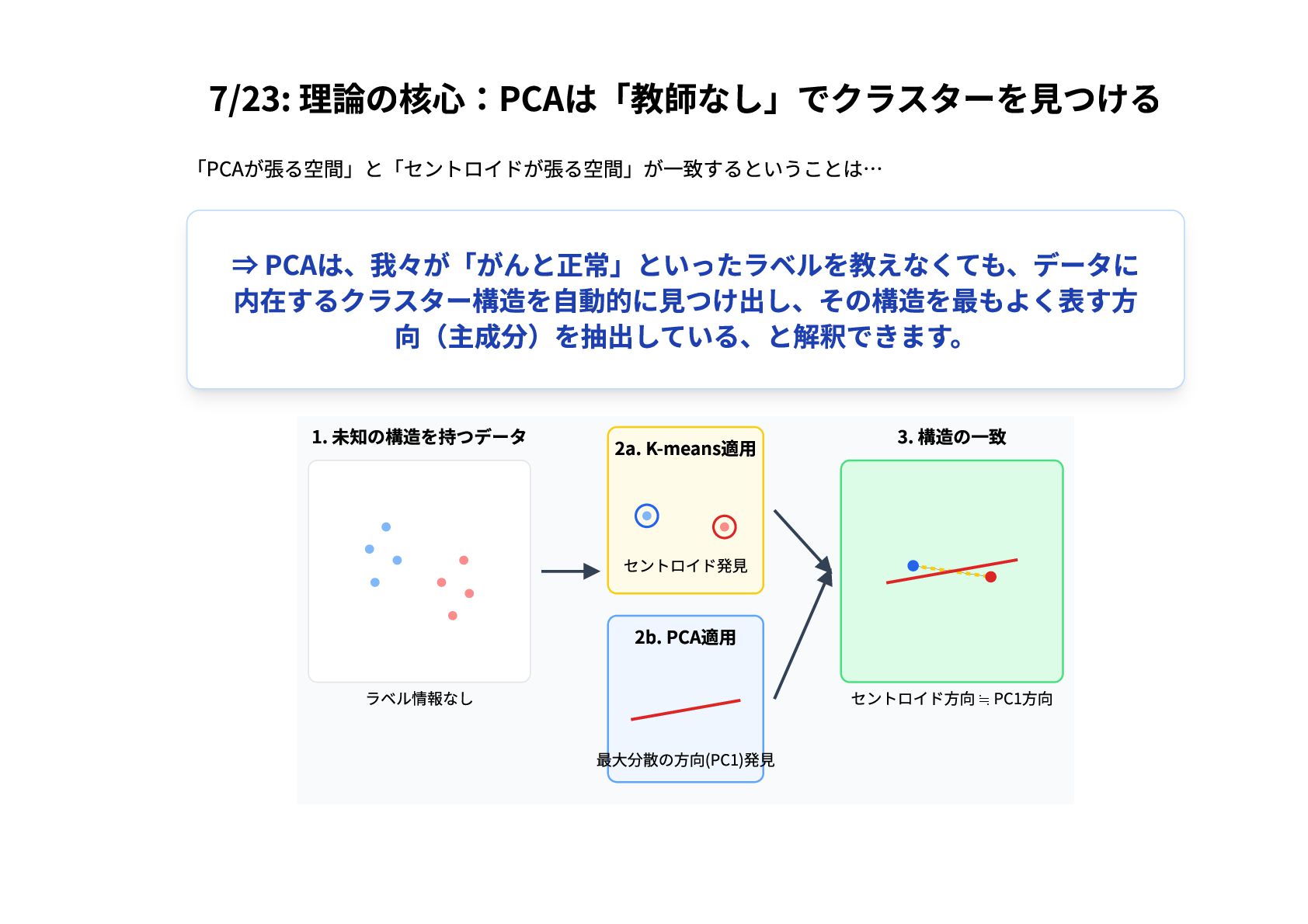
7/23: 理論の核心:PCAは「教師なし」でクラスターを見つける 「PCAが張る空間」と「セントロイドが張る空間」が一致するということは… ⇒ PCAは、我々が「がんと正常」といったラベルを教えなくても、データに 内在するクラスター構造を自動的に見つけ出し、その構造を最もよく表す方 向(主成分)を抽出している、と解釈できます。 1. 未知の構造を持つデータ ラベル情報なし
2a. K-means適用 セントロイド発見 2b. PCA適用 最大分散の方向(PC1)発見 3. 構造の一致 セントロイド方向 ≒ PC1方向
8/23: 本研究の仮説 以上の理論的背景から、本研究では以下の仮説を立てました。 仮説 PCA/TDベースの教師なし特徴抽出が成功する理由は、その手法が「がんと正常」のような生物学的に 意味のあるクラスターのセントロイド方向へデータを射影していることと等価だからである。 つまり、手法が見つけ出す「面白い射影方向」が、生物学的に重要な方向と一致している、という仮説 です。
9/23: 仮説の検証アプローチ この仮説を検証するため、2つの異なるアプローチの結果を比較します。 1. TDベース特徴抽出 (教師なし) TDを用いて、データ内在の構造(特異値ベクト ル)を純粋に抽出する。 2. 射影追跡
(PP) (教師あり) 意図的に「がん/正常」という正解ラベルの方向 にデータを射影する。 ⇒ もし仮説が正しければ、この2つの手法で選ばれた遺伝子は強く一致するはずです。
10/23: 使用データセット 検証には、過去にTDベース特徴抽出が有効性を示した3つの実際のデータセットを用いました。 データセット 1 & 2: 腎臓がん 腎臓がん組織と正常組織のmRNAおよびmiRNA発현 データ。
生物学的な違い:「がん vs 正常」 データセット 3: SARS-CoV-2 SARS-CoV-2ウイルスを感染させたヒト培養細胞の遺伝子発現データ。 生物学的な違い:「ウイルス感染 vs 非感染」
11/23: サンプルから遺伝子へ:特異値ベクトルの関係 これまでは、サンプルを区別する特異値ベクトル (v) に注目しました。では、遺伝子の重要度を示す特異値ベク トル (u) はどのように得られるのでしょうか? サンプルベクトル v
(サンプルを分類) × 遺伝子発現行列 X (N遺伝子 × Mサンプル) 遺伝子ベクトル u (遺伝子の寄与度) 元のデータ行列 X を介して、サンプルの特異値ベクトル v から遺伝子の特異値ベクトル u が計算されます。 (式: $u \propto Xv$ ) この関係により、サンプルを分類するパターンが、どの遺伝子によって駆動されているかを特定できます。
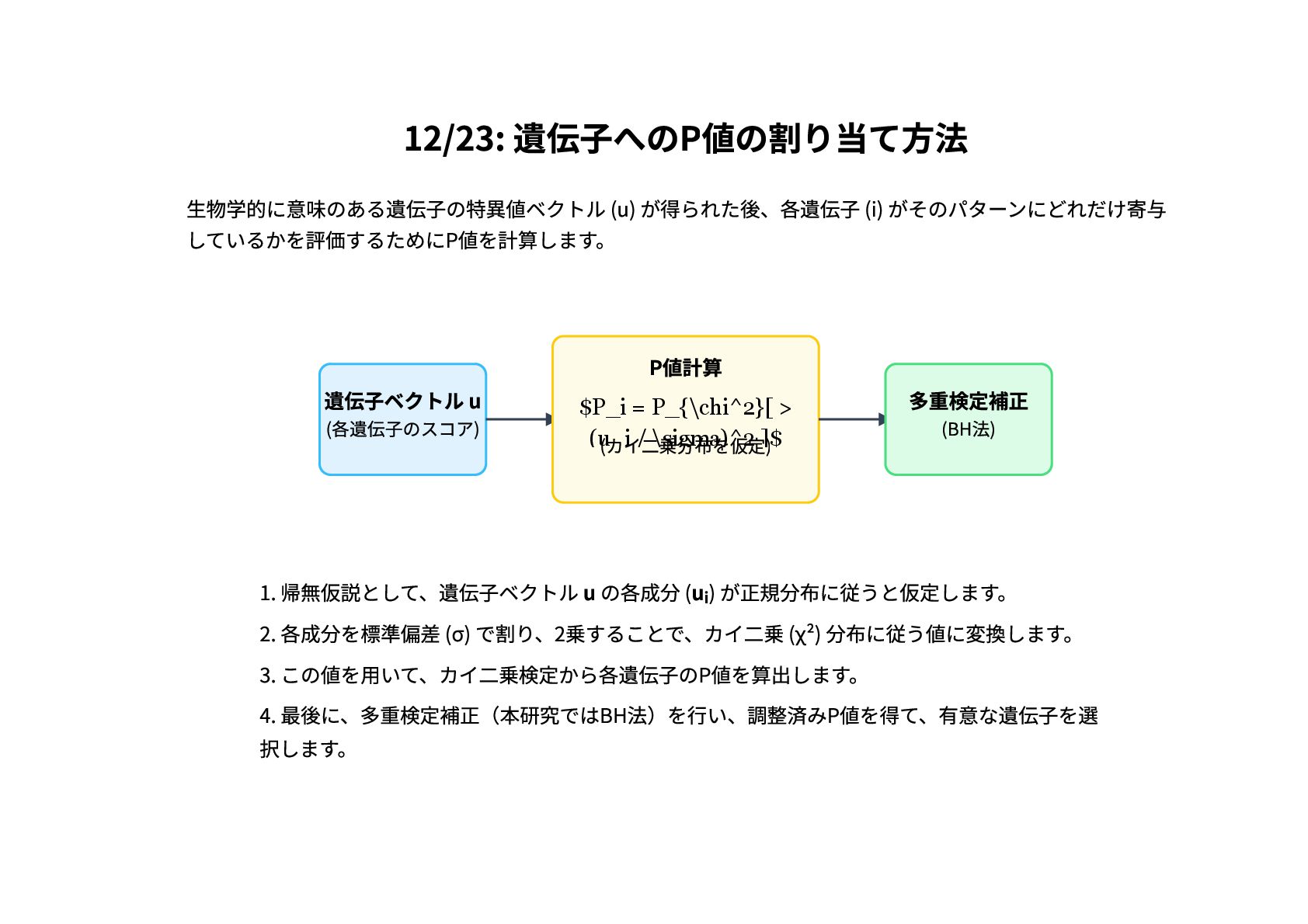
12/23: 遺伝子へのP値の割り当て方法 生物学的に意味のある遺伝子の特異値ベクトル (u) が得られた後、各遺伝子 (i) がそのパターンにどれだけ寄与 しているかを評価するためにP値を計算します。 遺伝子ベクトル u
(各遺伝子のスコア) P値計算 $P_i = P_{\chi^2}[ > (u i / \sigma)^2 ]$ (カイ二乗分布を仮定) 多重検定補正 (BH法) 1. 帰無仮説として、遺伝子ベクトル u の各成分 (u) が正規分布に従うと仮定します。 2. 各成分を標準偏差 (σ) で割り、2乗することで、カイ二乗 (χ²) 分布に従う値に変換します。 3. この値を用いて、カイ二乗検定から各遺伝子のP値を算出します。 4. 最後に、多重検定補正(本研究ではBH法)を行い、調整済みP値を得て、有意な遺伝子を選 択します。 i
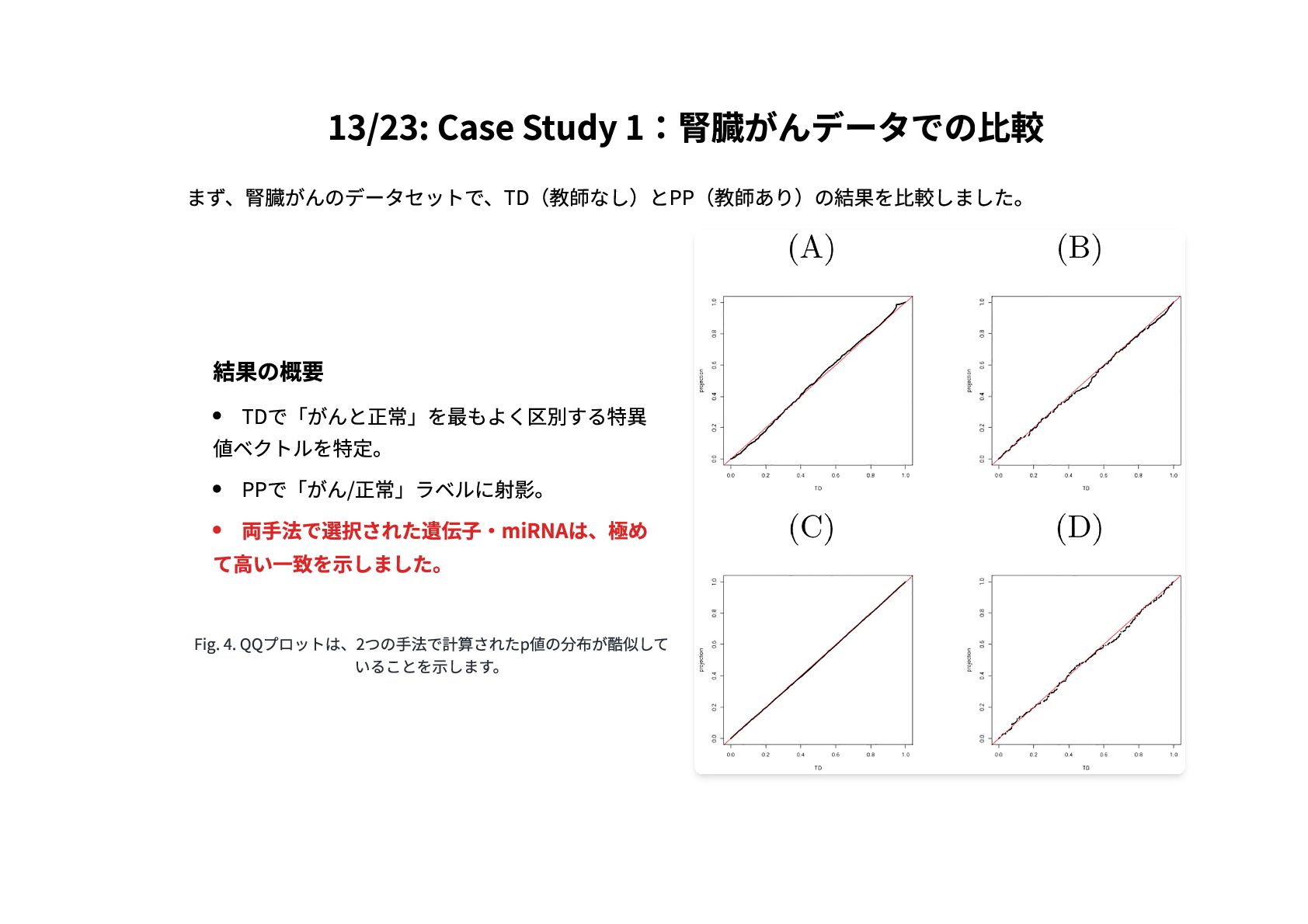
13/23: Case Study 1:腎臓がんデータでの比較 まず、腎臓がんのデータセットで、TD(教師なし)とPP(教師あり)の結果を比較しました。 結果の概要 TDで「がんと正常」を最もよく区別する特異 値ベクトルを特定。 PPで「がん/正常」ラベルに射影。 両手法で選択された遺伝子・miRNAは、極め
て高い一致を示しました。 Fig. 4. QQプロットは、2つの手法で計算されたp値の分布が酷似して いることを示します。
14/23: 腎臓がんデータでの一致度(表) 選択された遺伝子の一致度をクロス集計表で見てみましょう。(論文 Table 1, 2) mRNA (19,536遺伝子中) (p<0.01) PP
(選択) PP (非選択) TD (選択) 61 11 TD (非選択) 17 19,447 Fisher's exact test, p = 1.90 x 10⁻ ¹⁴⁹ miRNA (825 miRNA中) (p<0.01) PP (選択) PP (非選択) TD (選択) 11 0 TD (非選択) 2 812 Fisher's exact test, p = 2.76 x 10⁻ ²³ ⇒ この驚異的な一致は、TDが「がん/正常」という生物学的意味を持つ方向を的確に捉えていること を強く示唆します。
15/23: Case Study 2:SARS-CoV-2 データでの比較 次に、SARS-CoV-2感染細胞のデータセットでも同様の比較を行いました。(論文 Table 5) 結果の概要 TDで「ウイルス感染の有無」を区別する特異値ベクトルを特定し、163遺伝子を選択。
PPで「感染/非感染」ラベルに射影し、155遺伝子を選択。 腎臓がんデータと同様、両手法で選択された遺伝子は非常によく一致しました。 遺伝子クロス集計表 (21,897遺伝子中) (p<0.01) PP (選択) PP (非選択) TD (選択) 103 60 TD (非選択) 52 21,582 Fisher's exact test, p = 1.40 x 10⁻ ²⁴¹
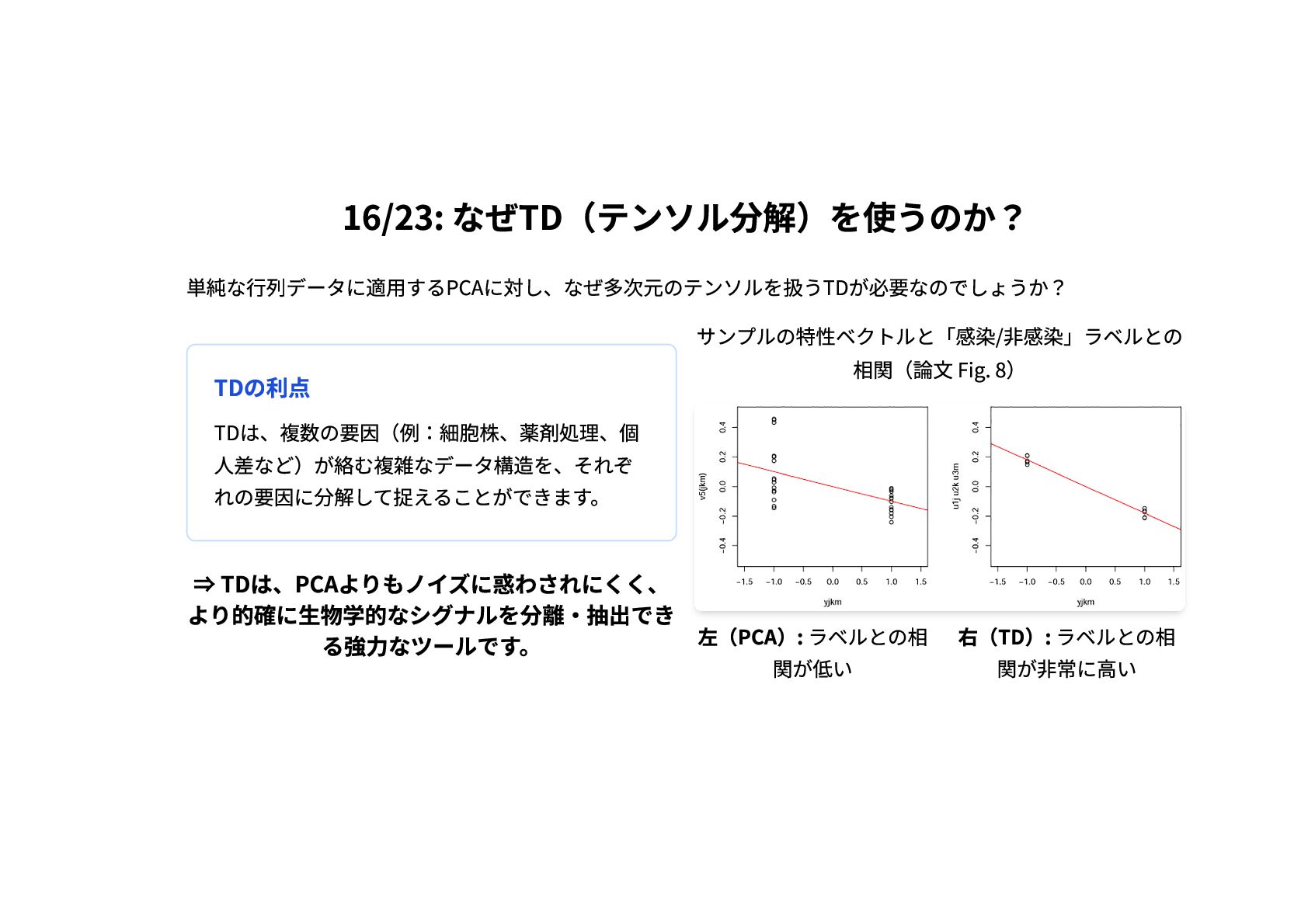
16/23: なぜTD(テンソル分解)を使うのか? 単純な行列データに適用するPCAに対し、なぜ多次元のテンソルを扱うTDが必要なのでしょうか? TDの利点 TDは、複数の要因(例:細胞株、薬剤処理、個 人差など)が絡む複雑なデータ構造を、それぞ れの要因に分解して捉えることができます。 ⇒ TDは、PCAよりもノイズに惑わされにくく、 より的確に生物学的なシグナルを分離・抽出でき
る強力なツールです。 サンプルの特性ベクトルと「感染/非感染」ラベルとの 相関(論文 Fig. 8) 左(PCA): ラベルとの相 関が低い 右(TD): ラベルとの相 関が非常に高い
17/23: 検証(1) - p値の閾値は妥当か? これまで経験的に「正規分布を仮定した調整済みp値 < 0.01」という基準が使われてきました。 しかし、この仮定と閾値は 本当に妥当なのでしょうか? この長年の疑問に答えるため、より信頼性の高いとされるシャッフリング法の結果と比較検証を行いました。
18/23: 検証(2) - シャッフリング法によるp値計算 シャッフリング法とは? 1. 各サンプル内で、遺伝子の発現データの並び順をランダムに入れ替える。 2. これにより、遺伝子と発現値の関連が破壊された「意味のない」データセットが 作られる。
3. このデータにTDを適用し、偶然得られるスコアの分布(帰無分布)を作成す る。 4. 実際のスコアが、この帰無分布の上位何%に位置するかを計算し、p値とする。 計算コストは非常に高いですが、データの分布に仮定を置かないため、頑健な方法とされています。
19/23: シャッフリング法によるp値分布の実例 シャッフリング法によって生成された帰無分布から計算されたp値の実際のヒストグラムです。 Fig.1: データセット1 (miRNA) Fig.2: データセット1 (mRNA) Fig.3:
データセット3 (遺伝子) パネル(A)ではp値が1付近に偏る傾向が見られますが、発現量の高い遺伝子に限定する(B)ことで、より一様な分布に近づきま す。
20/23: 検証(3) - 2つの基準の一致 結果 TDベース特徴抽出(調整済みp < 0.01)で選択された遺伝子群は、 シャッフリング法(調整済みp <
0.1)で選択された遺伝子群と、 全てのデータセットで非常によく一致しました。 ⇒ 経験的に使われてきた簡易な基準が、頑健な手法の結果とよく相関しており、実用上有効であるこ とが初めて裏付けられました。
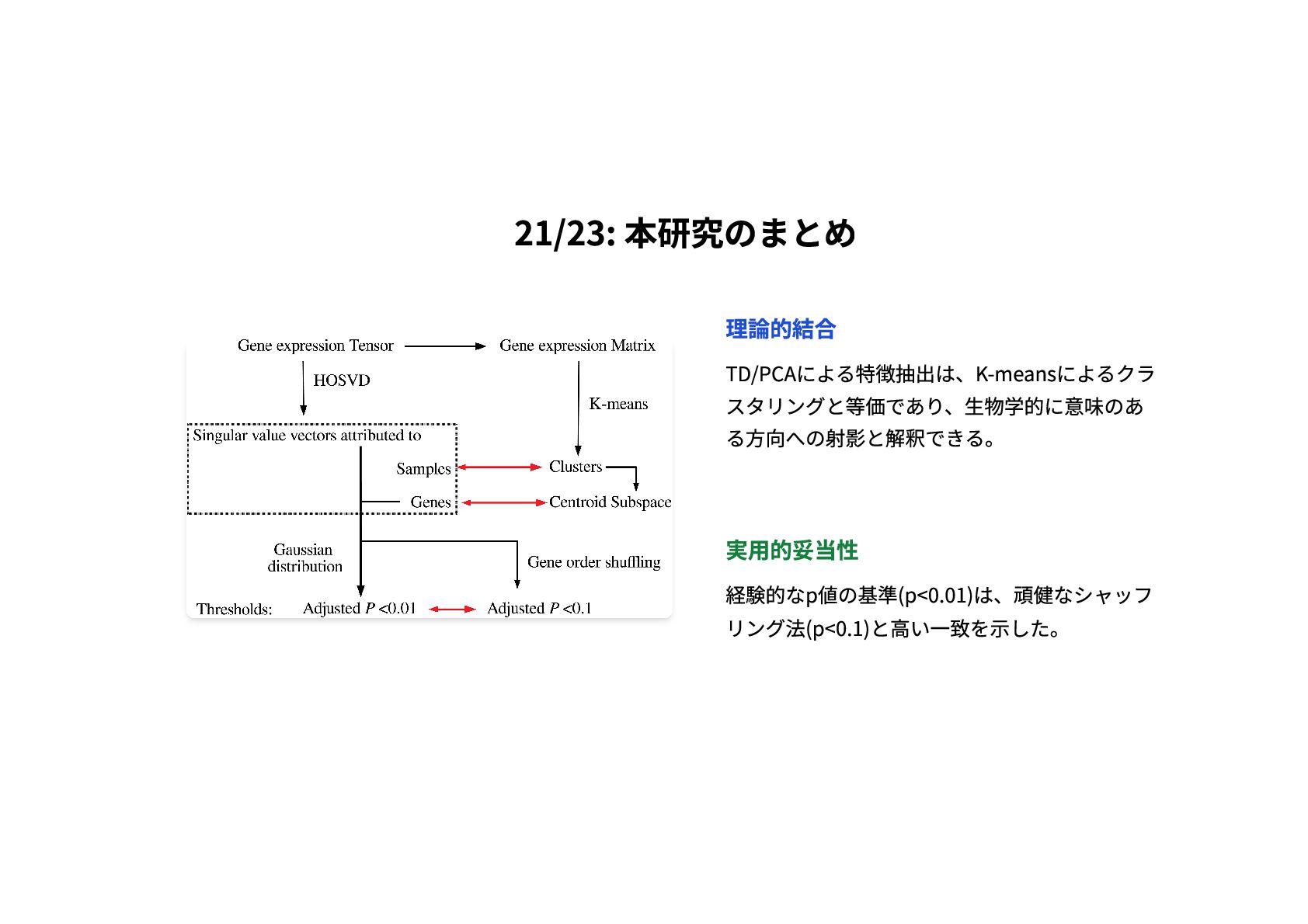
21/23: 本研究のまとめ 理論的結合 TD/PCAによる特徴抽出は、K-meansによるクラ スタリングと等価であり、生物学的に意味のあ る方向への射影と解釈できる。 実用的妥当性 経験的なp値の基準(p<0.01)は、頑健なシャッフ リング法(p<0.1)と高い一致を示した。
22/23: 結論 本研究は、ゲノム解析における特徴選択ツールとして、PCAおよびTDベース教師なし特徴抽 出の有効性を、射影という観点から理論的に初めて合理化しました。 これらの手法は、サンプル数が少ない高次元データ($N \gg M$)から、生物学的に意味のあ る特徴量を抽出するための、強力かつ理論的に裏付けられたアプローチです。 研究者は、データに隠された本質的な生物学的パターンを発見するために、これらの手法を 自信を持って活用することができます。
23/23: 今後の展望と謝辞 今後の展望 理論の拡張: 3つ以上のクラスターが存在す る場合や、連続的な変化(時間経過など)を捉 える場合の理論拡張。 応用範囲の拡大: マルチオミクスデータの統 合解析へのより広範な応用。個別化医療に向け
たバイオマーカー探索の加速。 謝辞 共同研究者: Dr. Turki Turki 研究費支援: 日本学術振興会(JSPS)科研費 ご清聴ありがとうございました。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}