This presentation gives an a short introduction about our experience deploying large scale genomic pipelines with Nextflow and AWS Batch cloud service.
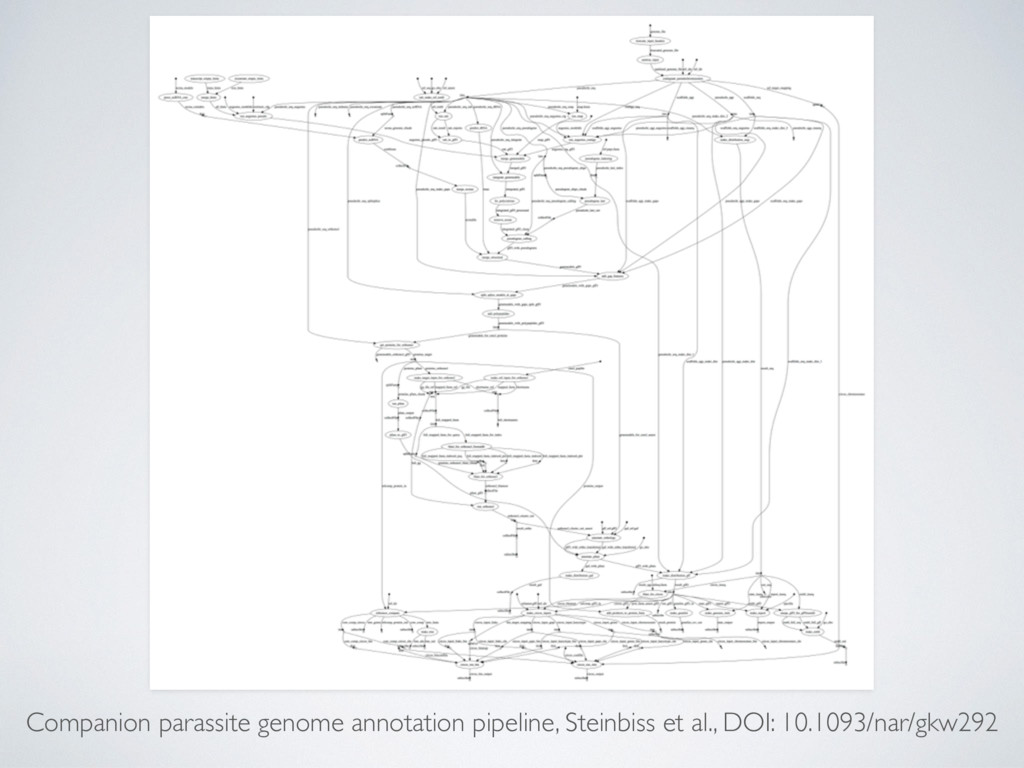
(large) genomic datasets • Mash-up of many different tools and scripts • Embarrassingly parallelisation, can spawn 100s-100k jobs over distributed cluster • Complex dependency trees and configuration → very fragile ecosystem
batch fashion (ie. asynchronous) • It manages the provisioning and scaling of the cluster • It provides the concept of queue • A job is a container (cool!)
including the job script • Upload the Docker image in a public registry • Create a job template referencing the upload image • Submit the job execution with the AWS command line tool
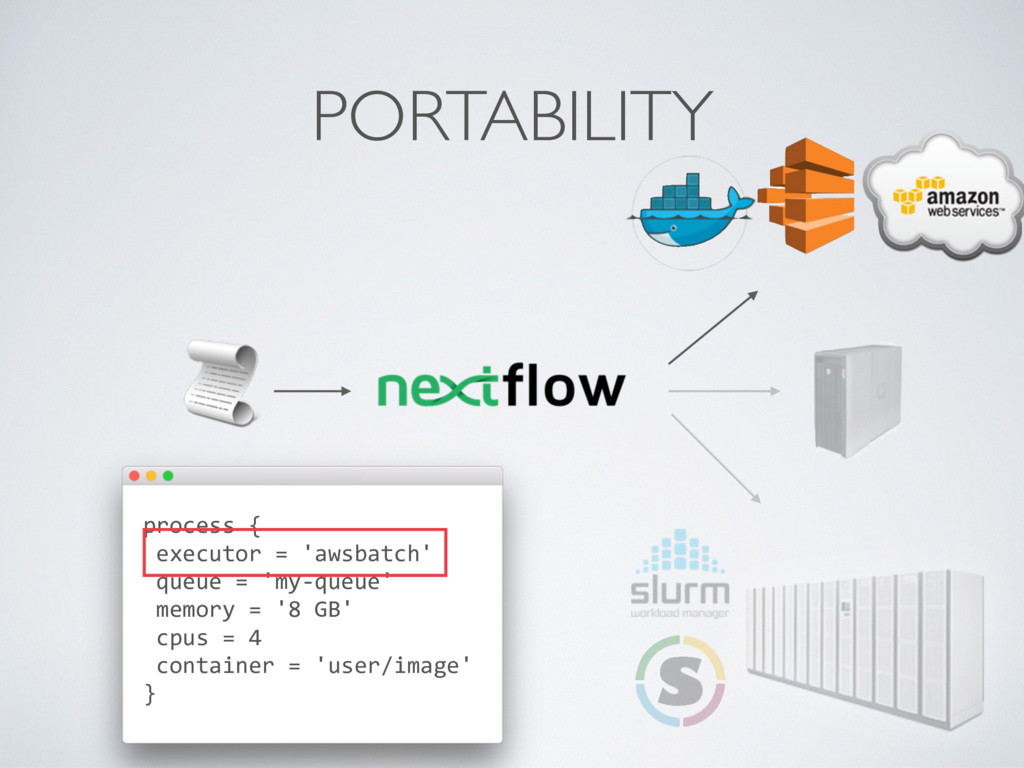
output uploads reduce the workflow portability • Custom container images (ideally we would like to use community container images eg. BioContainers) • Orchestrating big real-world workflows can be challenging
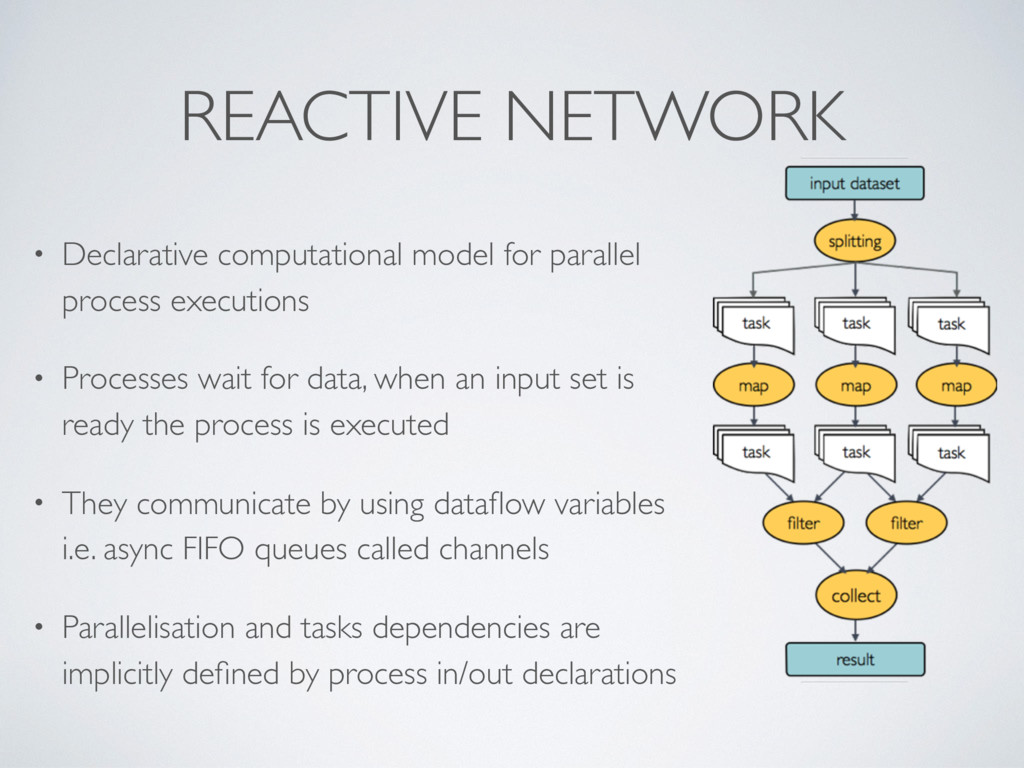
• Processes wait for data, when an input set is ready the process is executed • They communicate by using dataflow variables i.e. async FIFO queues called channels • Parallelisation and tasks dependencies are implicitly defined by process in/out declarations
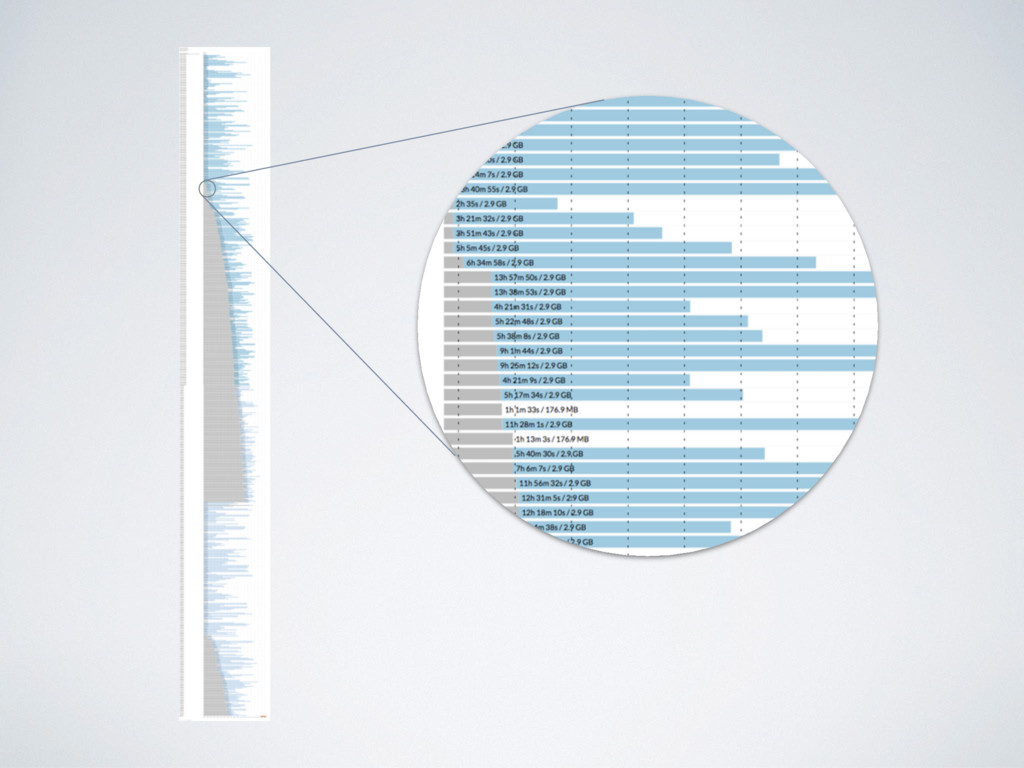
samples take from Encode project • 753 Jobs • ~65 i3.xlarge spot instances • ~23 h wall-time time ~4'850 CPU-hours * https://github.com/nextflow-io/rnaseq-encode-nf
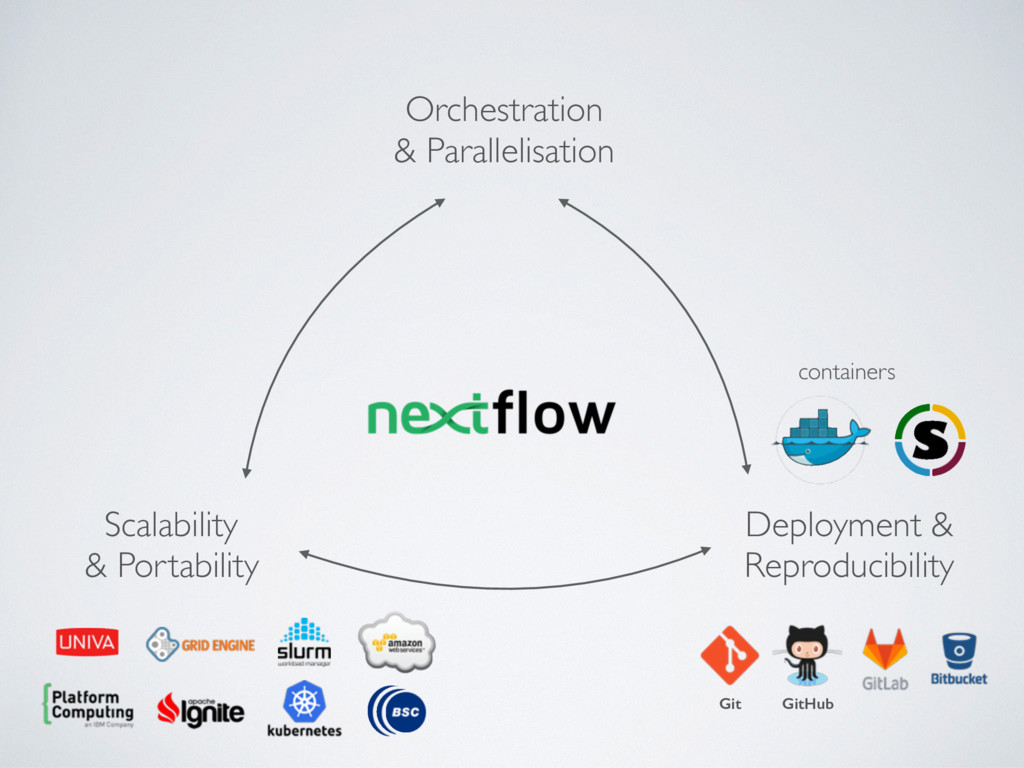
computing environment for containerised workloads • Delegating the cluster provisioning is a big plus • Choose carefully the size of EBS storage • Nextflow enables the seamless deployment of scalable and portable computational workflows
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}