Dataflow oriented bioinformatics pipelines with Nextflow
I introduce the Dataflow programming model. How it can be used to deal with the increasing complexity of bioinformatics pipelines, and how the Nextflow framework addresses many problems common in other approaches.
manipulation • No dependencies on external servers • It enables fast prototyping and experiments multiple alternative easily • Linux is the integration layer in the Bioinformatics domains
on the target platform • As script get bigger as it becomes fragile/ un-readable/hard to modify • In large script tasks inputs/outputs tends to be not clearly defined
scale properly with big data • Very different parallelization strategies and implementations • Shared memory (process, thread) • Message passing (MPI, actors) • Distributed computation • Hardware parallelization (GPU) • In general provide a too low level abstraction and requires specific API skills • Introduce hard dependencies to specific platform/framework
Originates in research for reactive system to monitor and control industrial processes • All tasks are parallel and form a process network • Tasks communicate through channels (non-blocking unidirectional FIFO queue) • A task is executed when all its inputs are bound • The synchronization is implicitly defined by tasks inputs/ outputs declaration 1. G. Kahn, “The Semantics of a Simple Language for Parallel Programming,” Proc. of the IFIP Congress 74, North-Holland Publishing Co., 1974
• It is executed as soon as declared inputs are available • It is inherently parallel • Each tasks is executed in its own private directory • Produced outputs trigger the execution of downstream tasks
different platforms by a simple definition into the configuration file • Local processes (Java threads) • Resource managers (SGE, LSF, SLURM, etc) • Cloud (Amazon Ec2, Google, etc)
gracefully and reports the error cause • Easy debugging, each tasks can be executed separately • When fixed, the execution can be resumed from the failure point
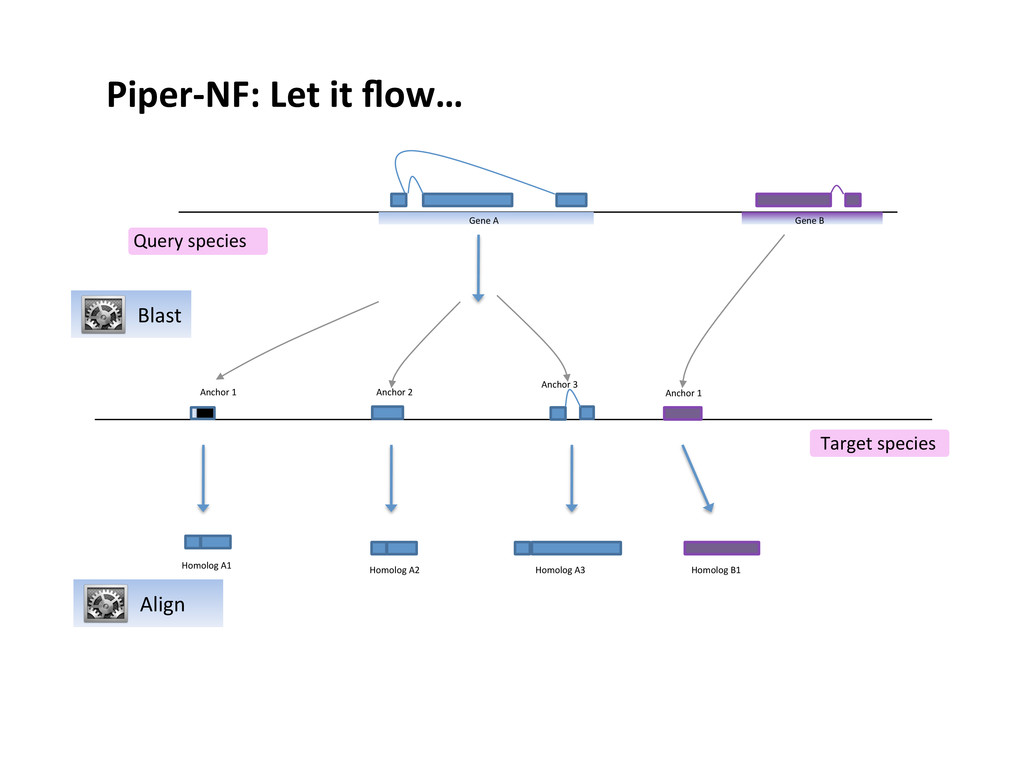
mapping of long non- coding RNAs • ~ 10'000 lines of Perl code • run a single computer (single process) or cluster through SGE • ~ 70% of the code deals with parameters handling, file splitting, jobs parallelization, synchronization, etc. • very inefficient parallelization
Parallelization depends on number of genomes (1 genomes no parallel execution) • A specific resource manages technology (qsub) is hard-coded into the pipeline • If it crash, you lost days of computation
of code vs. 10'000 legacy version • Much easier to write, test and to maintain • Implementation platform agnostic • Parallelized splitting by query and genome • Fine grain control on parallelization • Greatly improved tasks “interleaving”
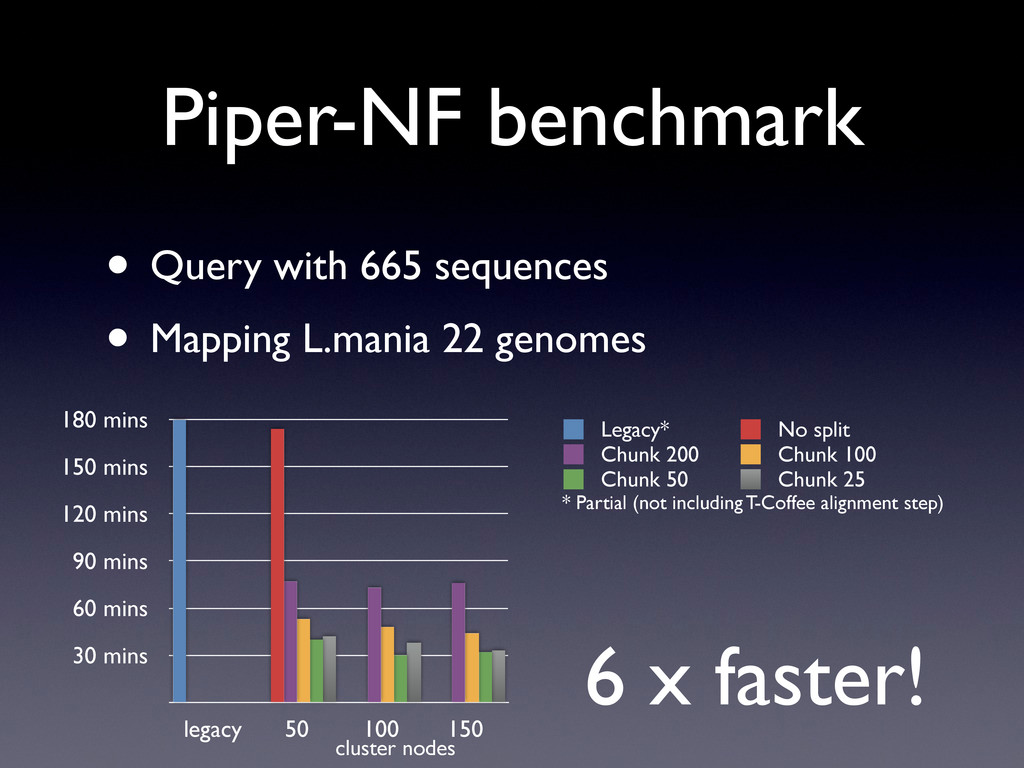
22 genomes 30 mins 60 mins 90 mins 120 mins 150 mins 180 mins legacy 50 100 150 Legacy* No split Chunk 200 Chunk 100 Chunk 50 Chunk 25 cluster nodes * Partial (not including T-Coffee alignment step) 6 x faster!
• Enhance syntax to make more expressive • Advanced profiling (Paraver) • Improve grid support (SLURM, LSF, DRMAA, etc) • Integrate the cloud (Amazon, DNAnexus) • Add health monitoring and automatic fail-over
run • Pipeline functional logic is decoupled by the actual execution layer (local/grid/cloud/?) • Reuse existing code/scripts • Parallelism is defined implicitly by tasks inputs/ outputs declarations • On task error, stops gently reporting the error, and resume from failed point
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Parallel BLAST example query = file(args[0]) DB = "$HOME/blast-‐db/pdb/pdb" seq](https://files.speakerdeck.com/presentations/0693d740bca101300f863e4d25b30b1b/slide_12.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}