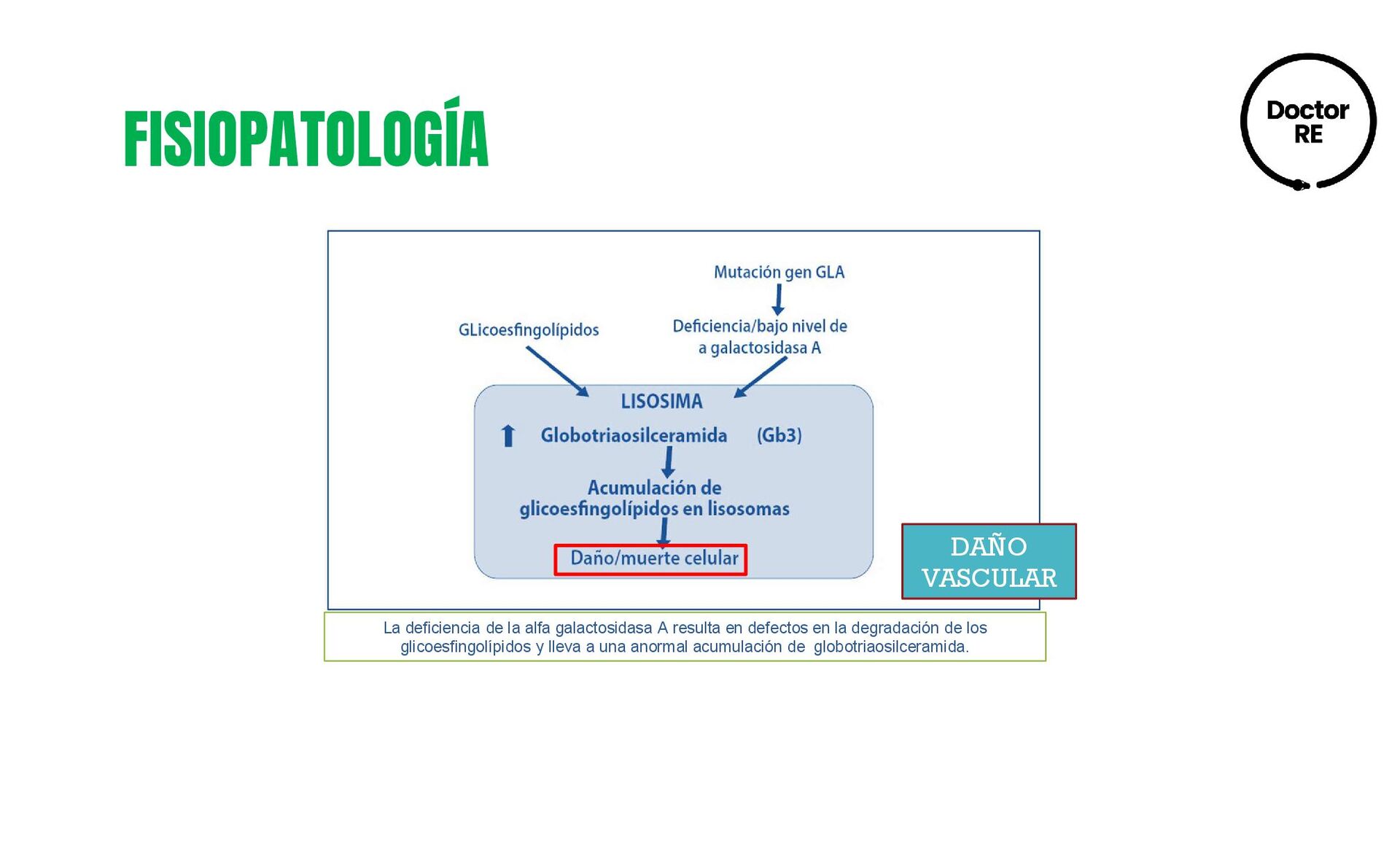
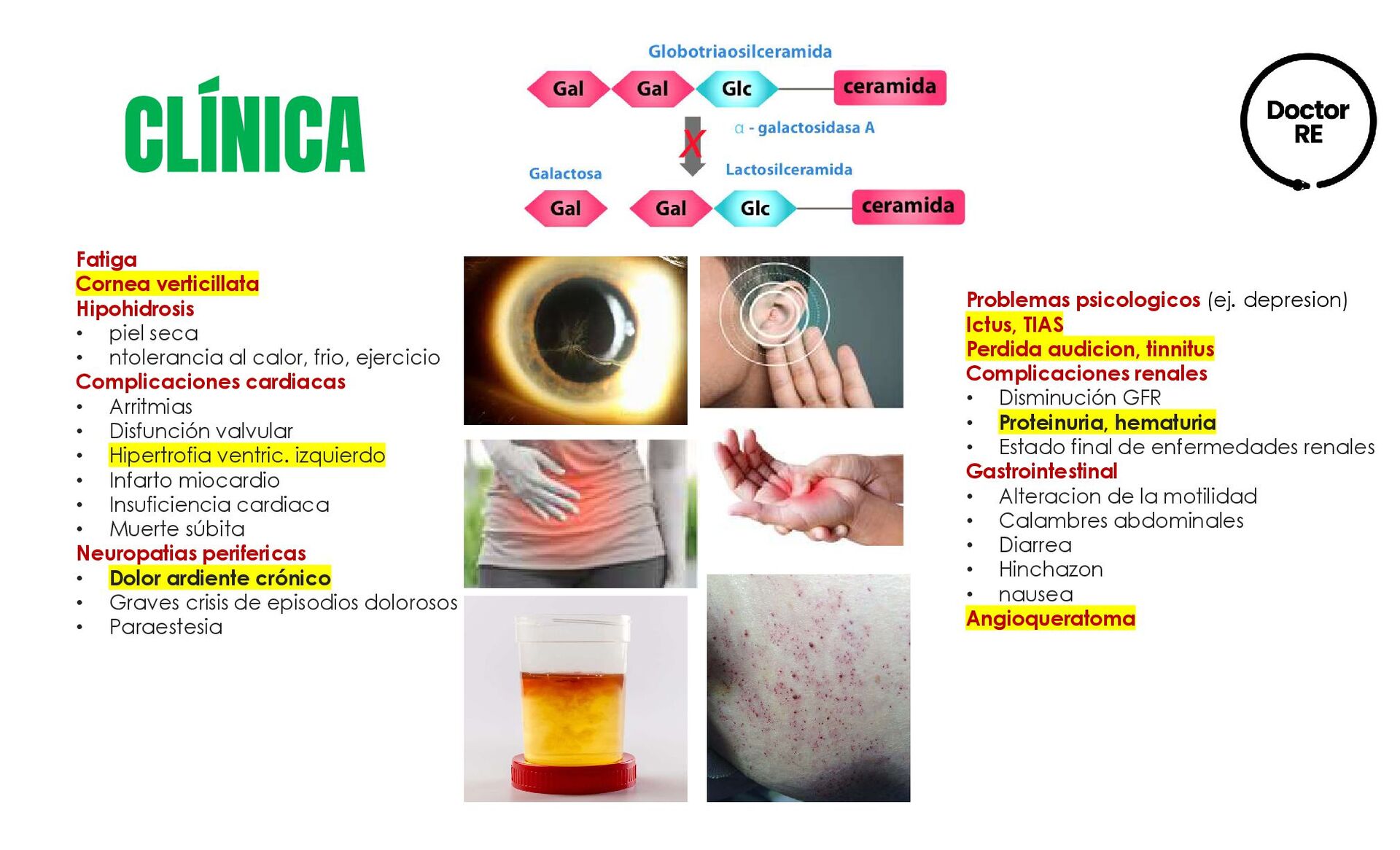
neuropático, dolores crónicos en las articulaciones, anomalías oftalmológicas, hipoacusia, hipersensibilidad al calor y el frío, trastornos gastrointestinales, letargo y fatiga, angioqueratomas, primeras anomalías renales y cardiacas. Edad adulta temprana (17-30 años). • Otros angioqueratomas, linfedemas en MP, riñones: proteinuria e IR avanzada • Corazón: miocardiopatía hipertrófica e hipertrofia de ventrículo izquierdo, angina de pecho, arritmias • SNC: ataque isquémico transitorio, infarto cerebral, depresión. Edad adulta avanzada (>30 años). • Insuficiencia cardiaca, trastornos del ritmo cardiaco • Recidiva de AIT e infarto cerebral • Insuficiencia renal, necesidad de diálisis
{kind=link}
{kind=link}
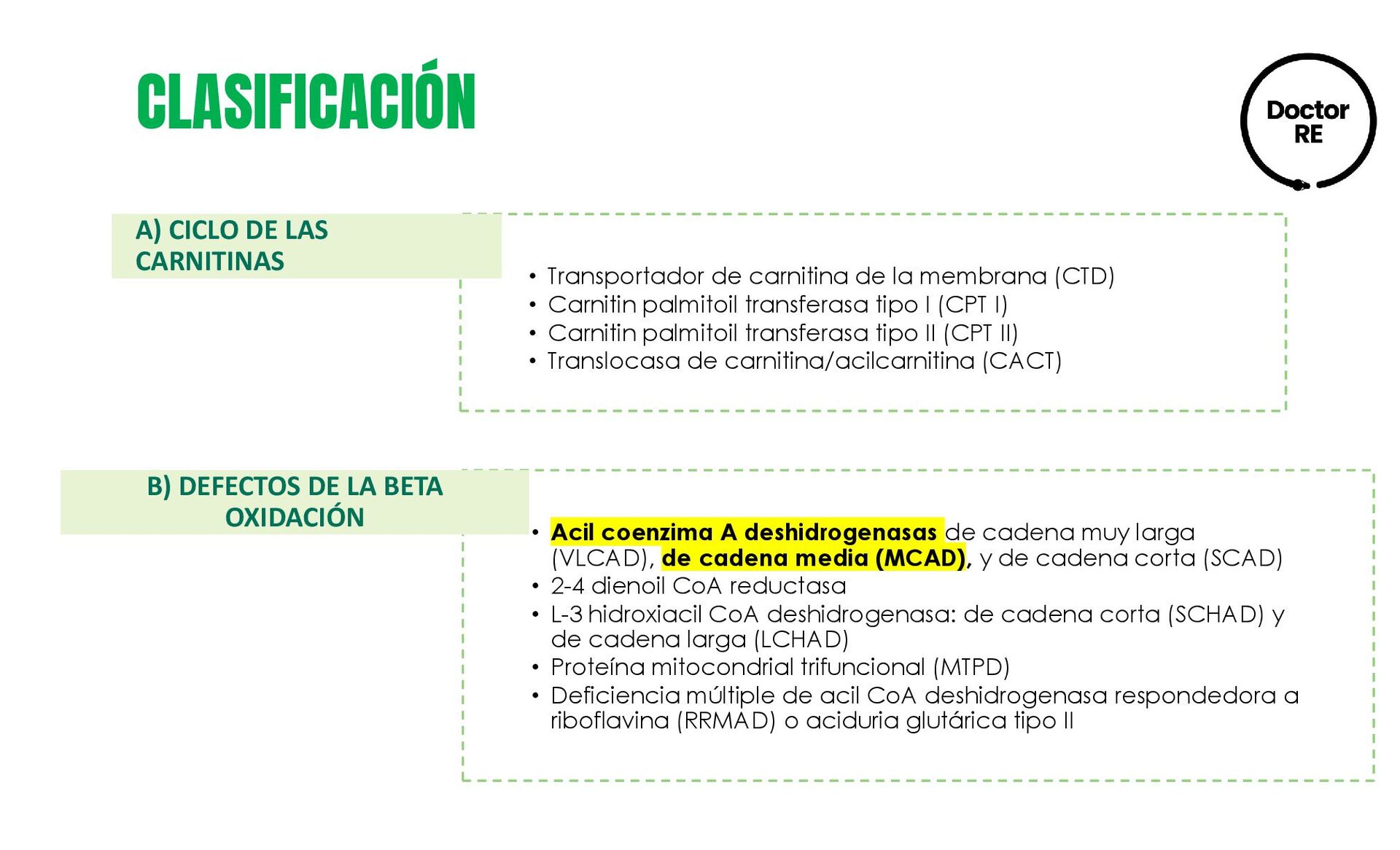{kind=link}
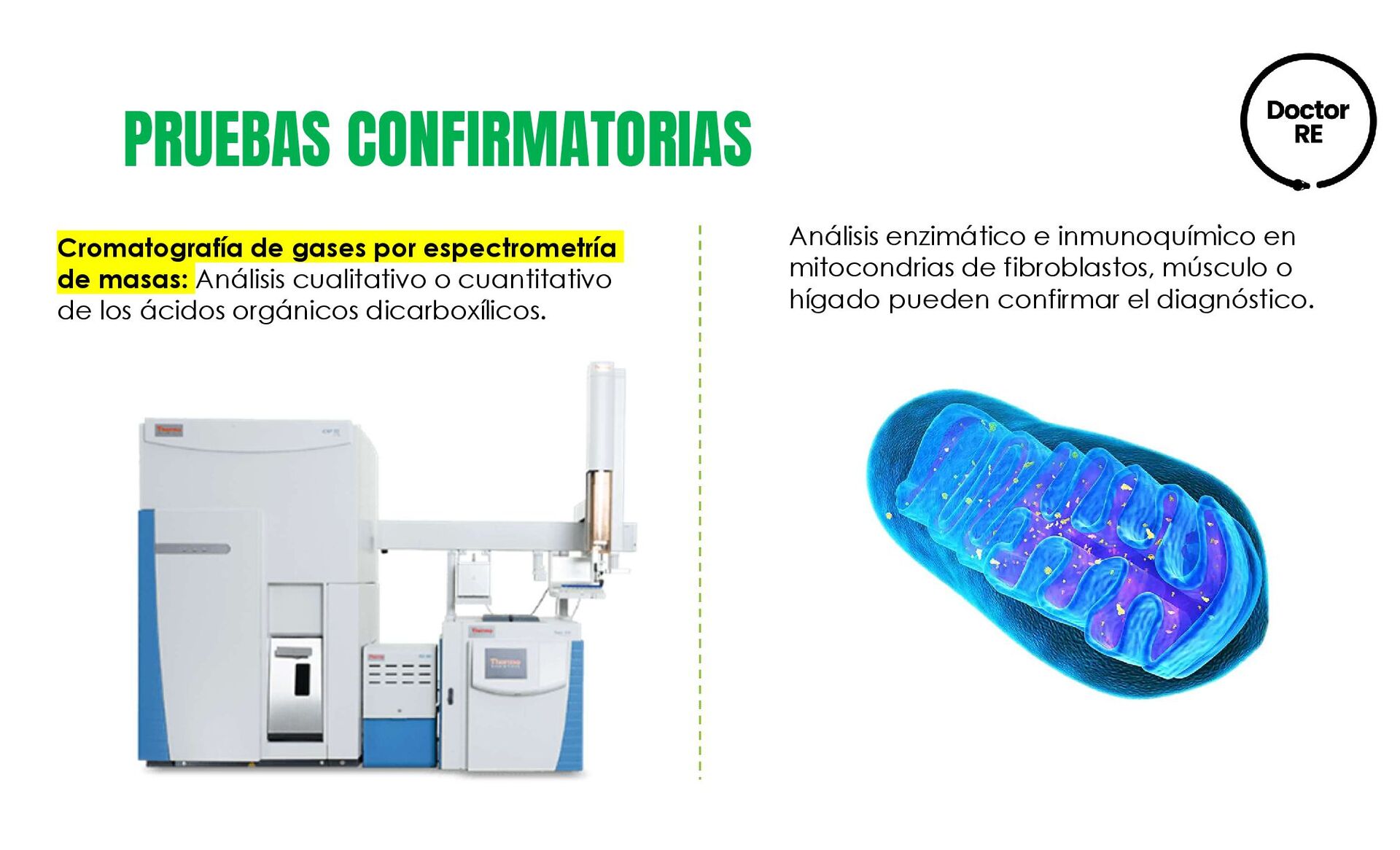{kind=link}
{kind=link}
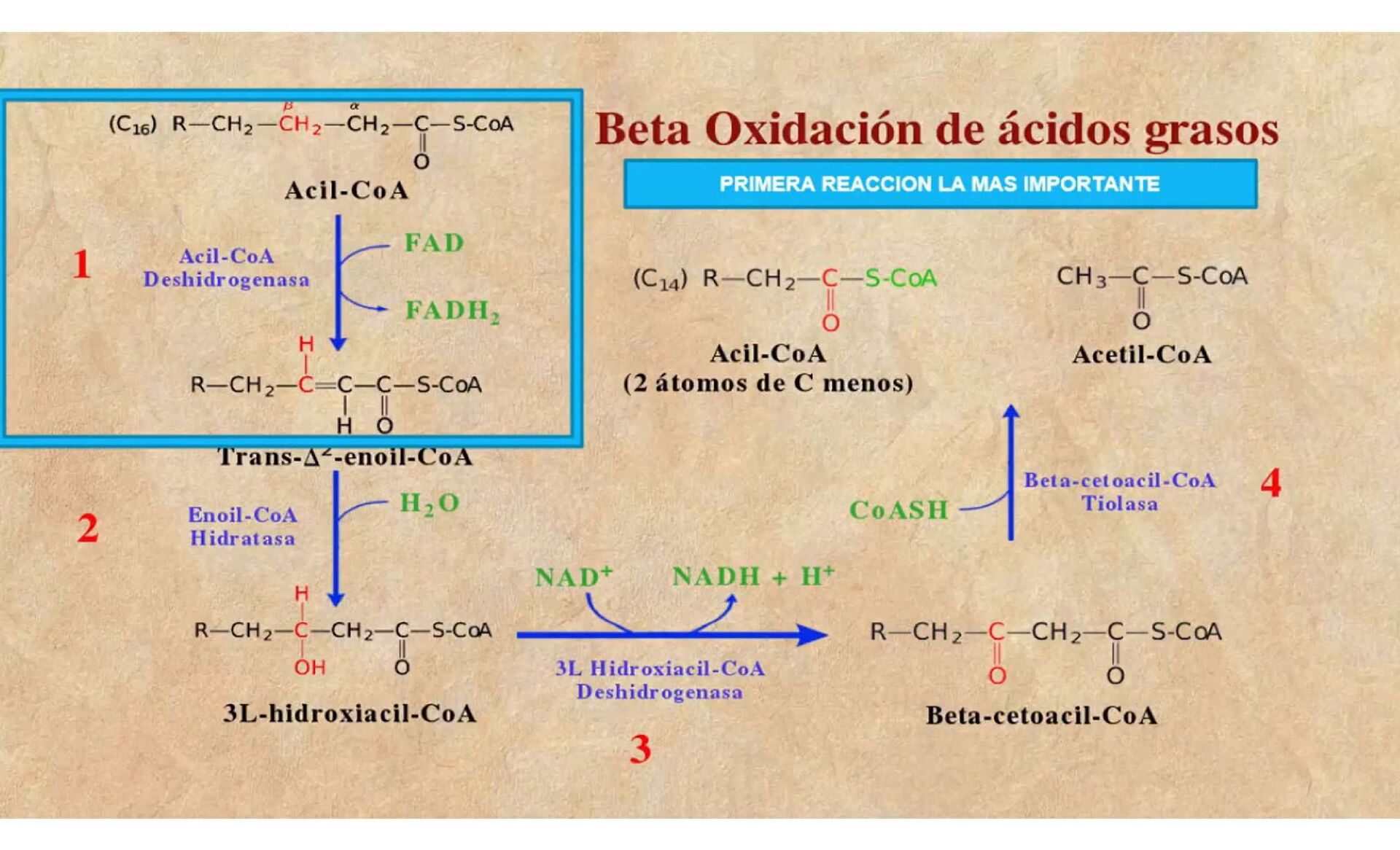{kind=link}
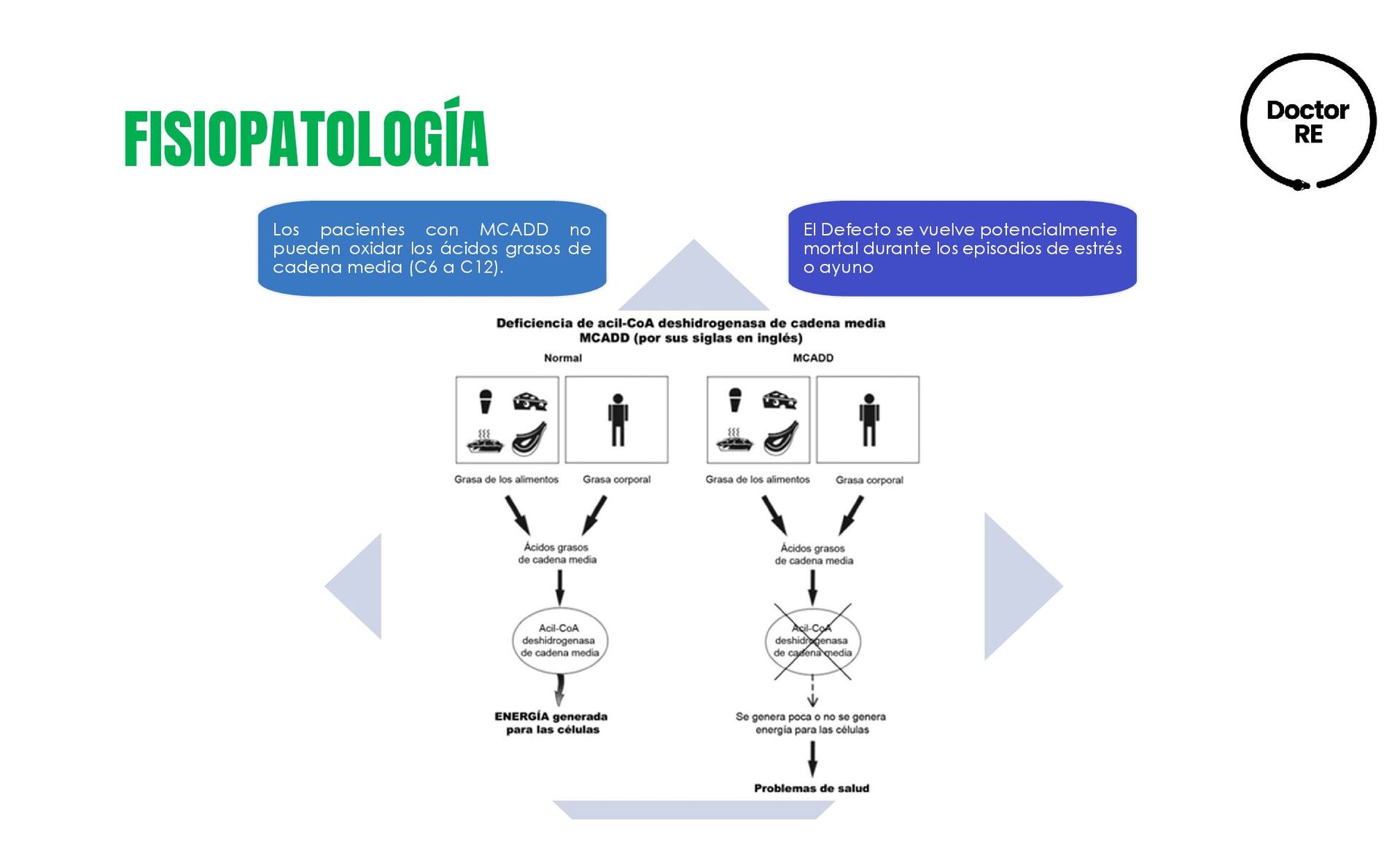{kind=link}
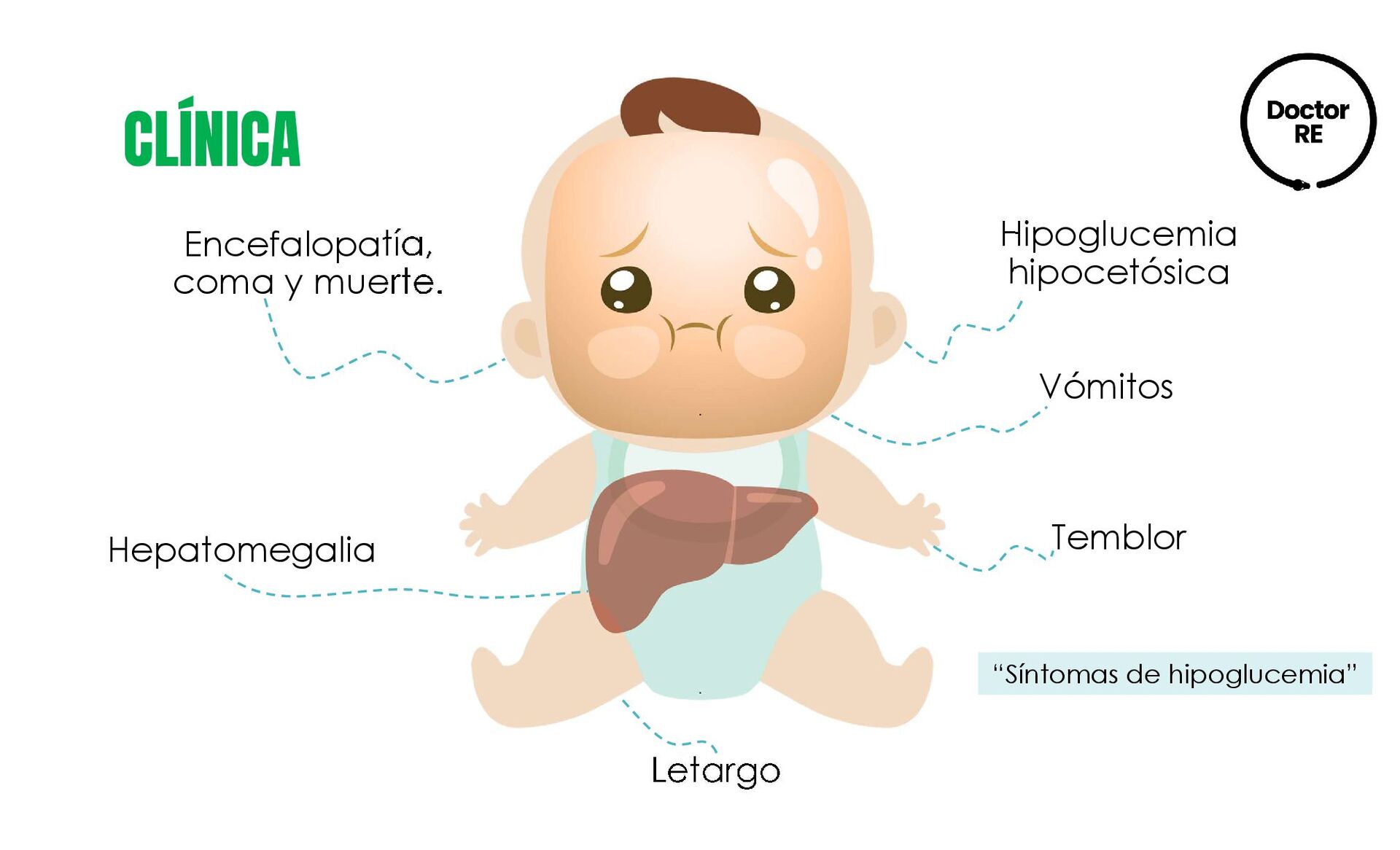{kind=link}
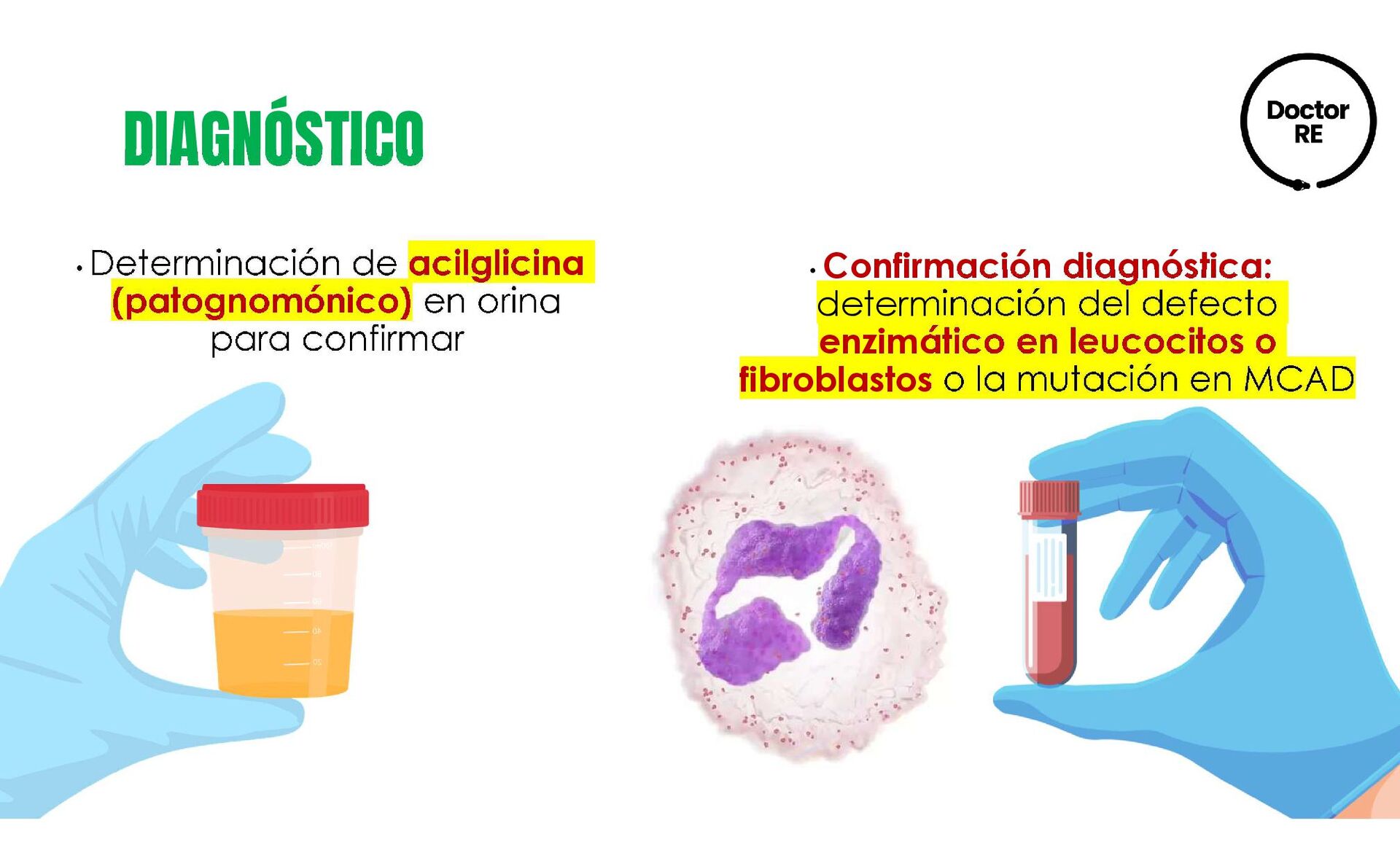{kind=link}
{kind=link}
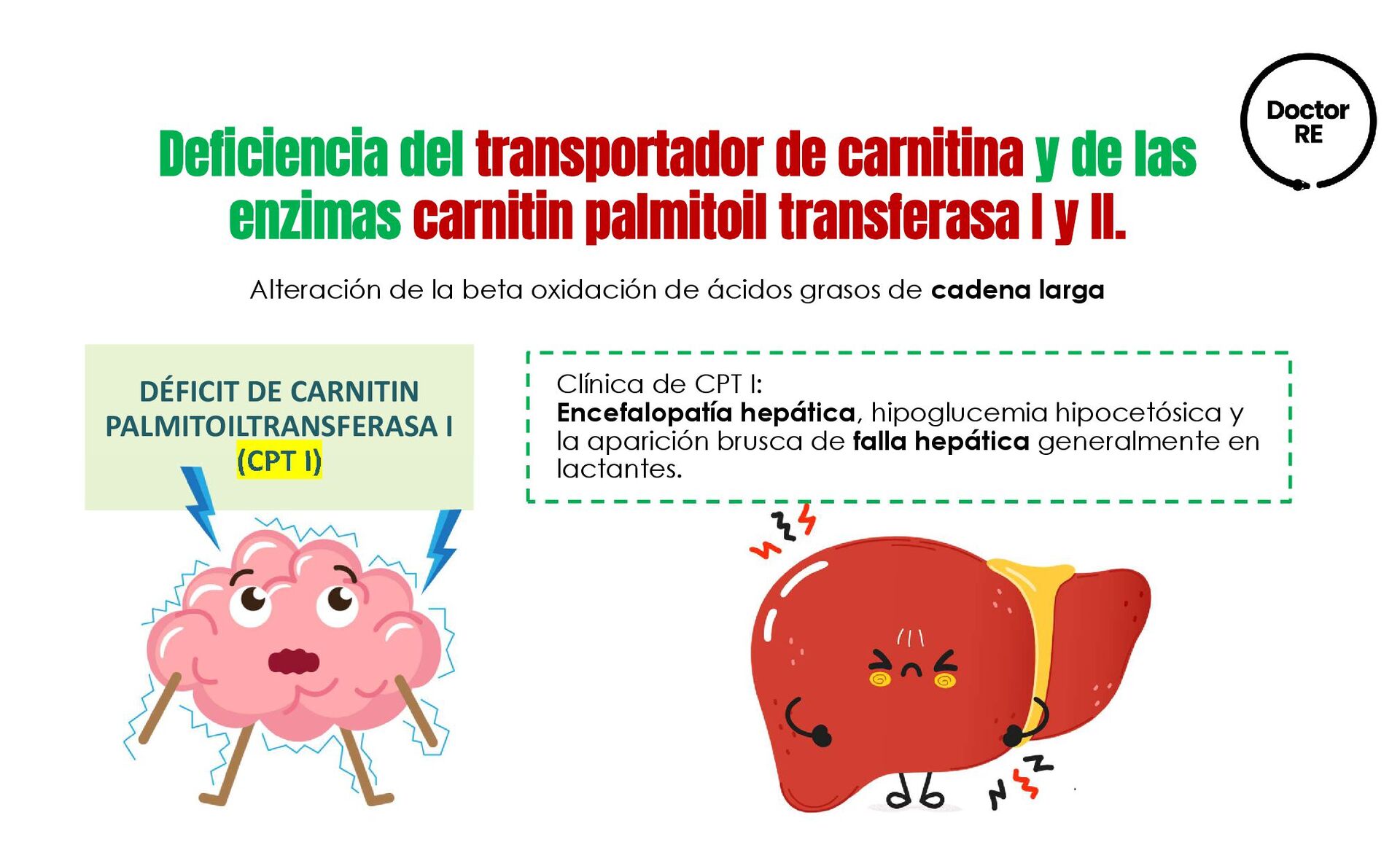{kind=link}
{kind=link}
{kind=link}
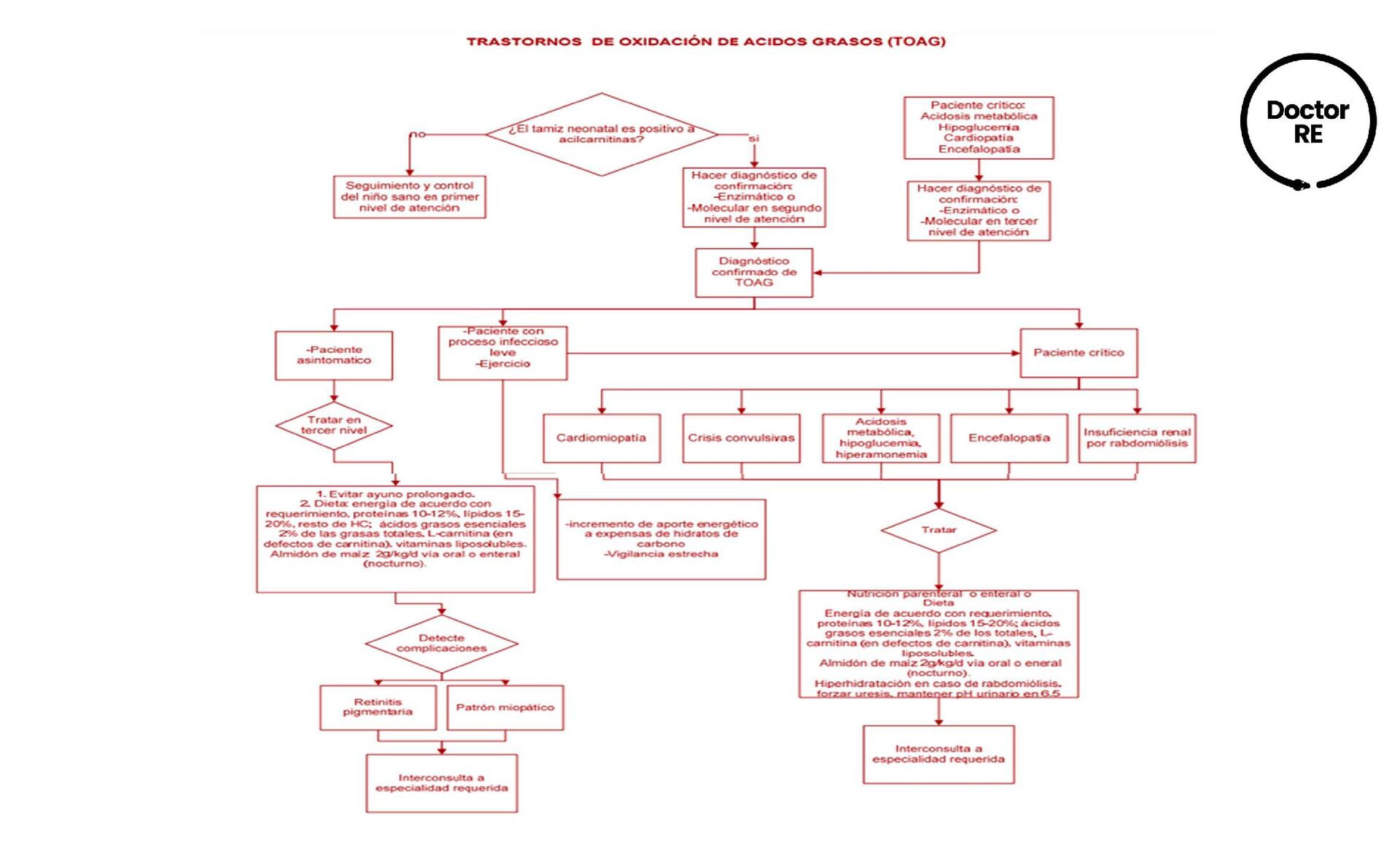{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
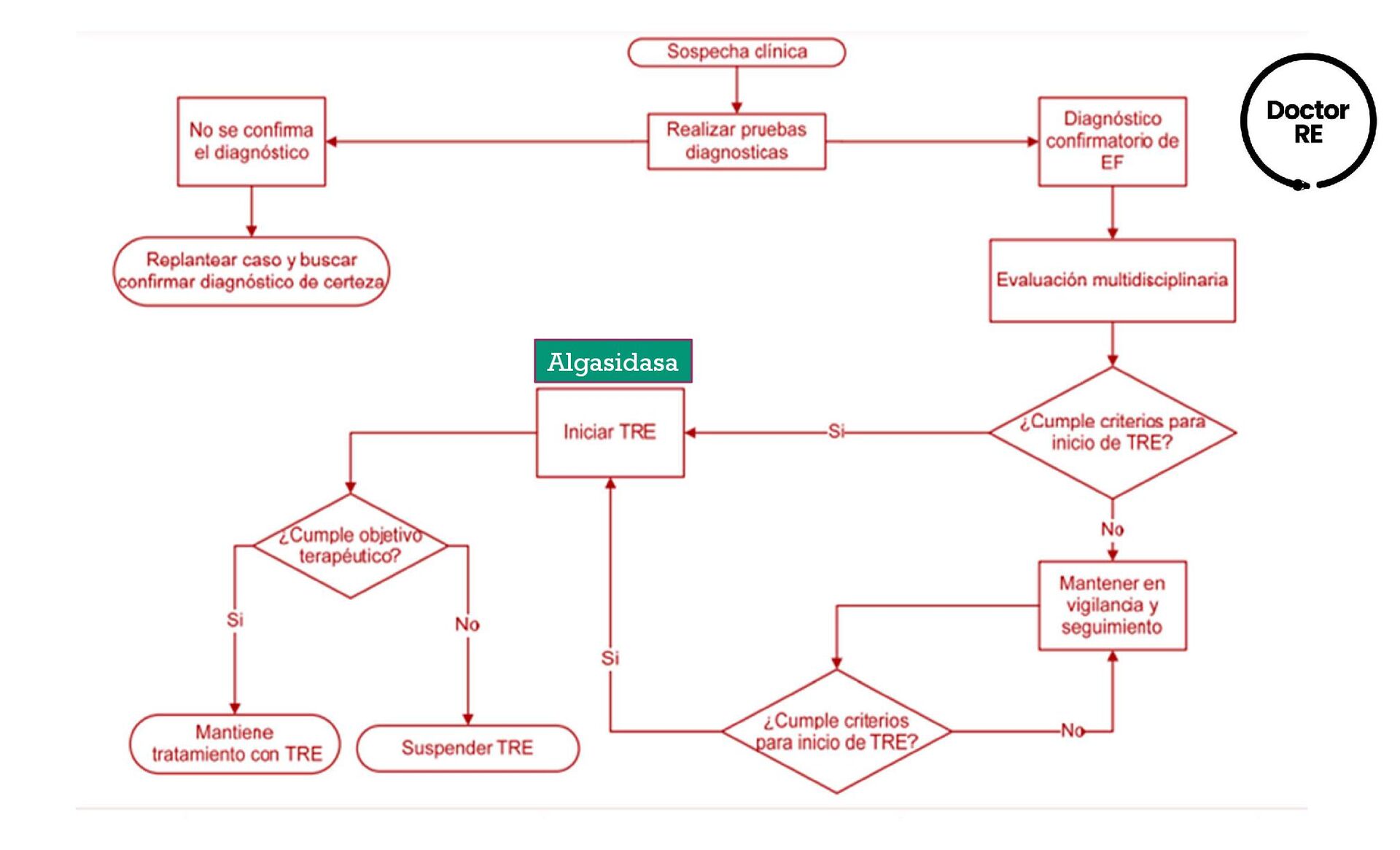{kind=link}
{kind=link}
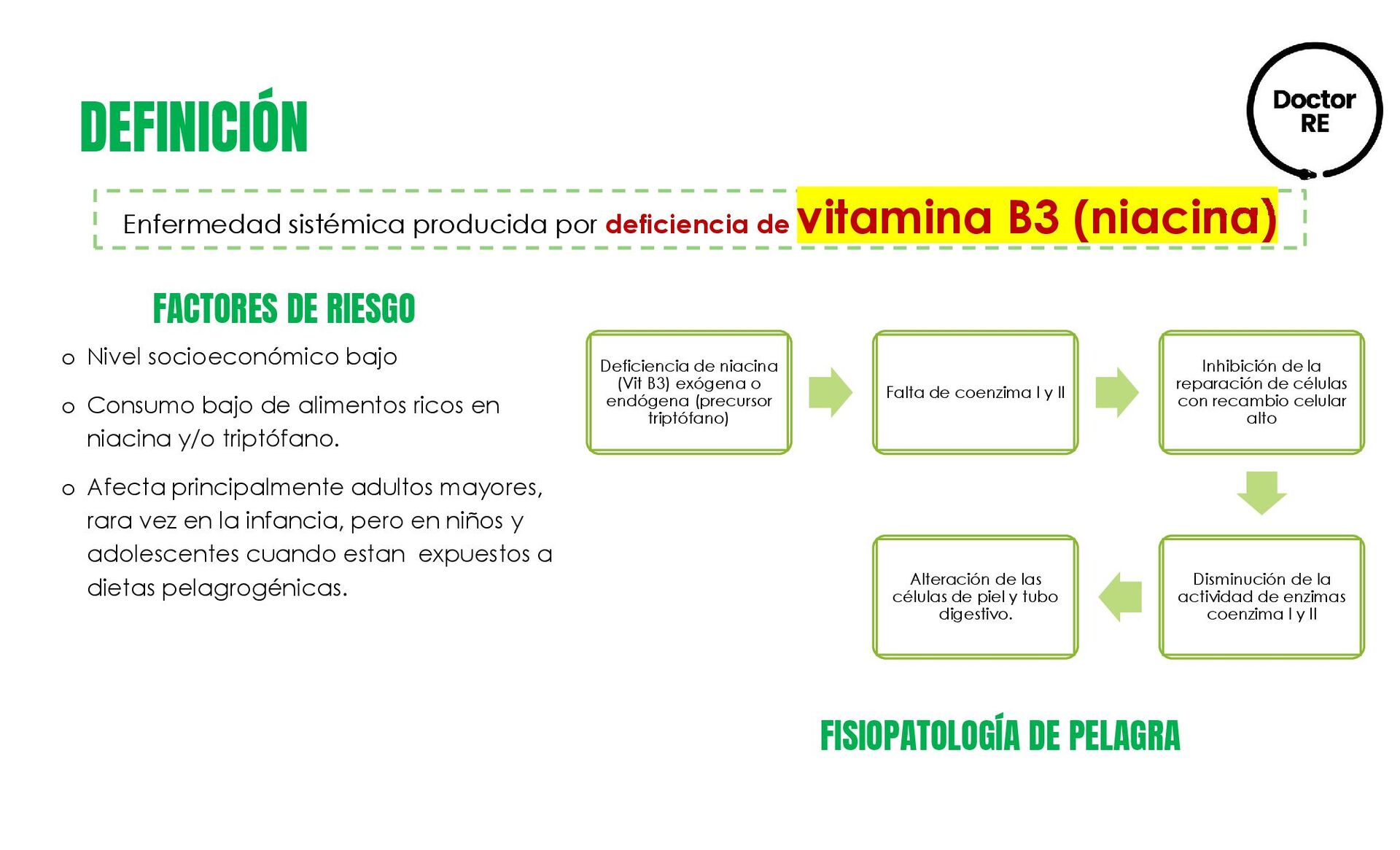{kind=link}
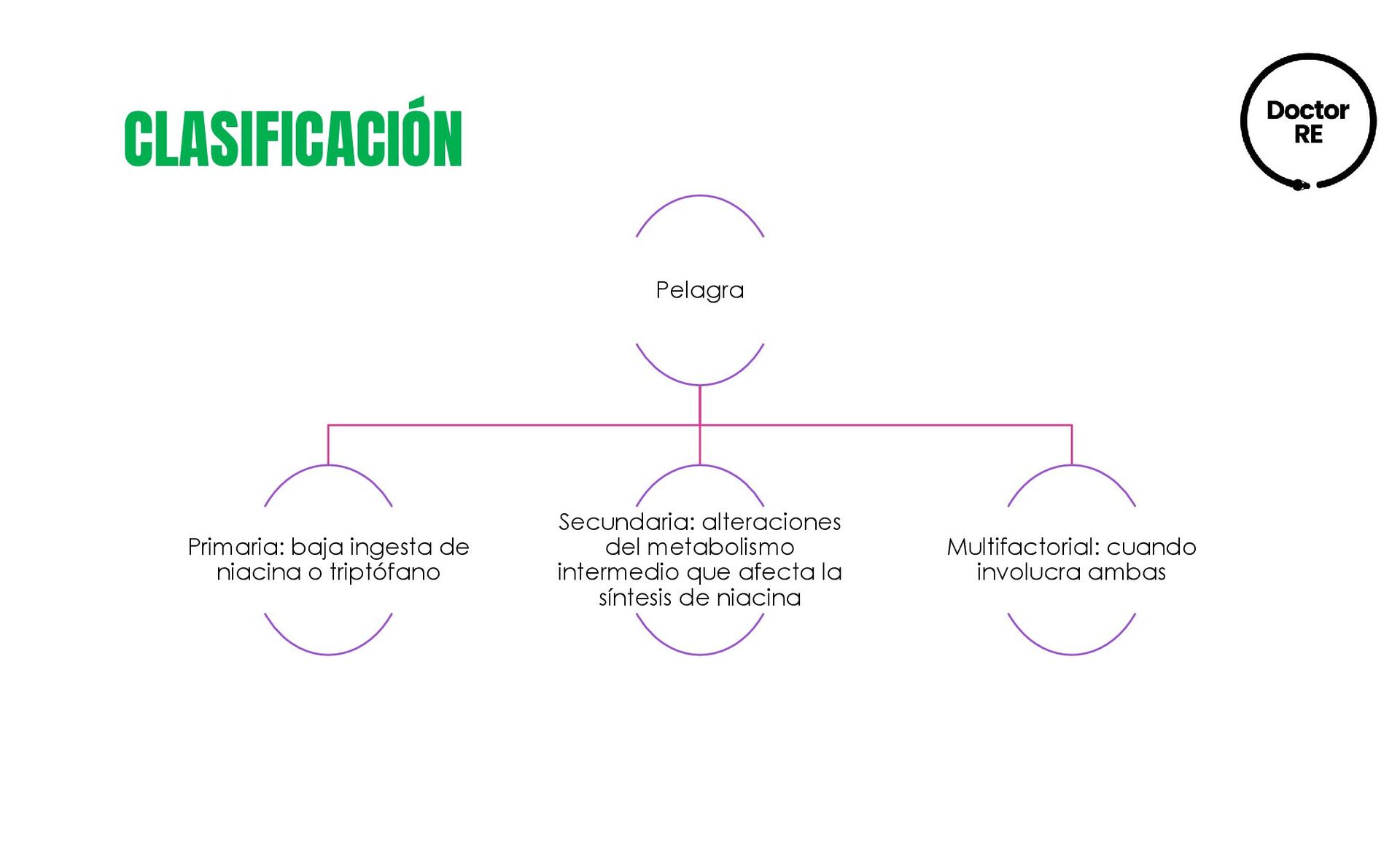{kind=link}
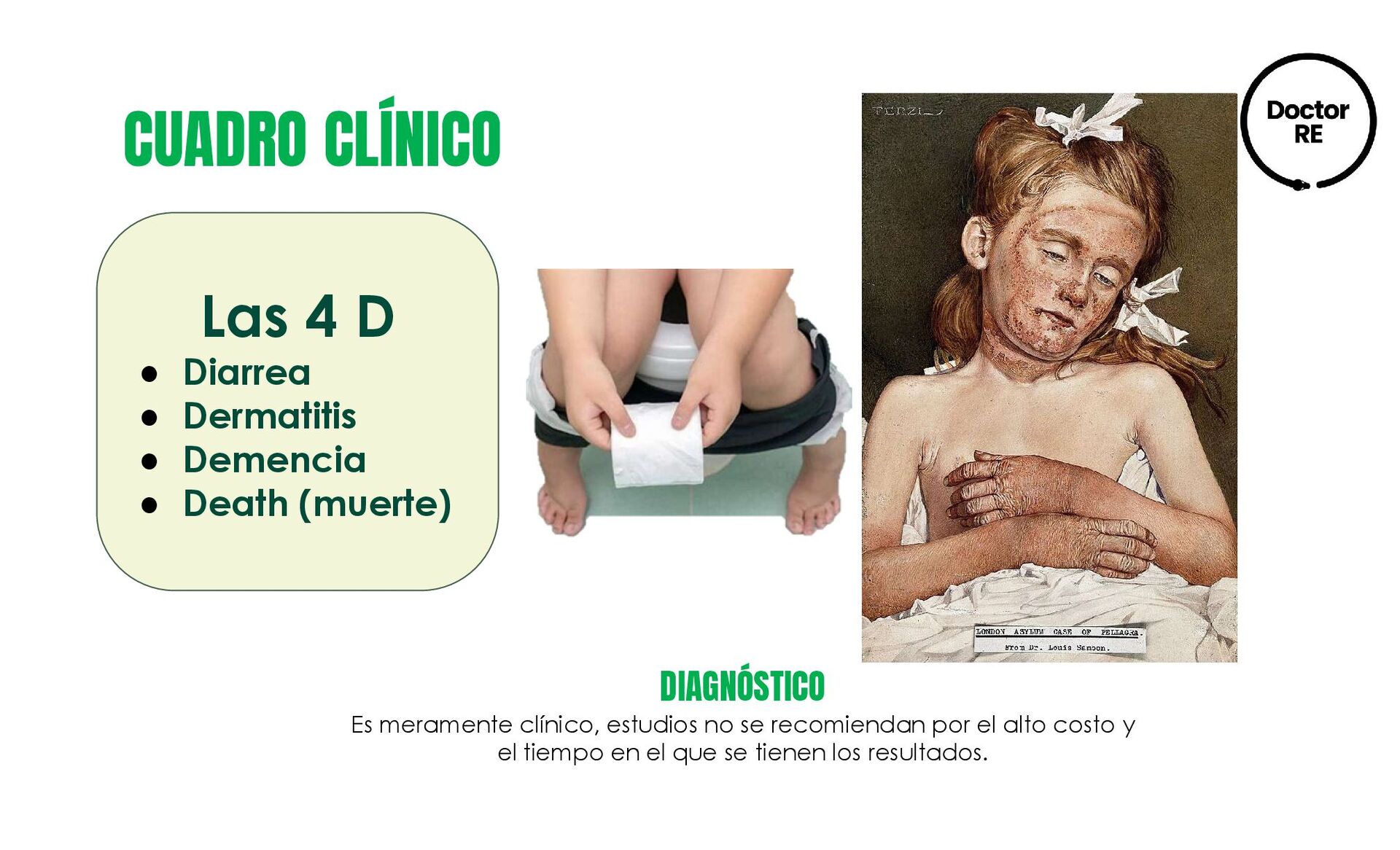{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}