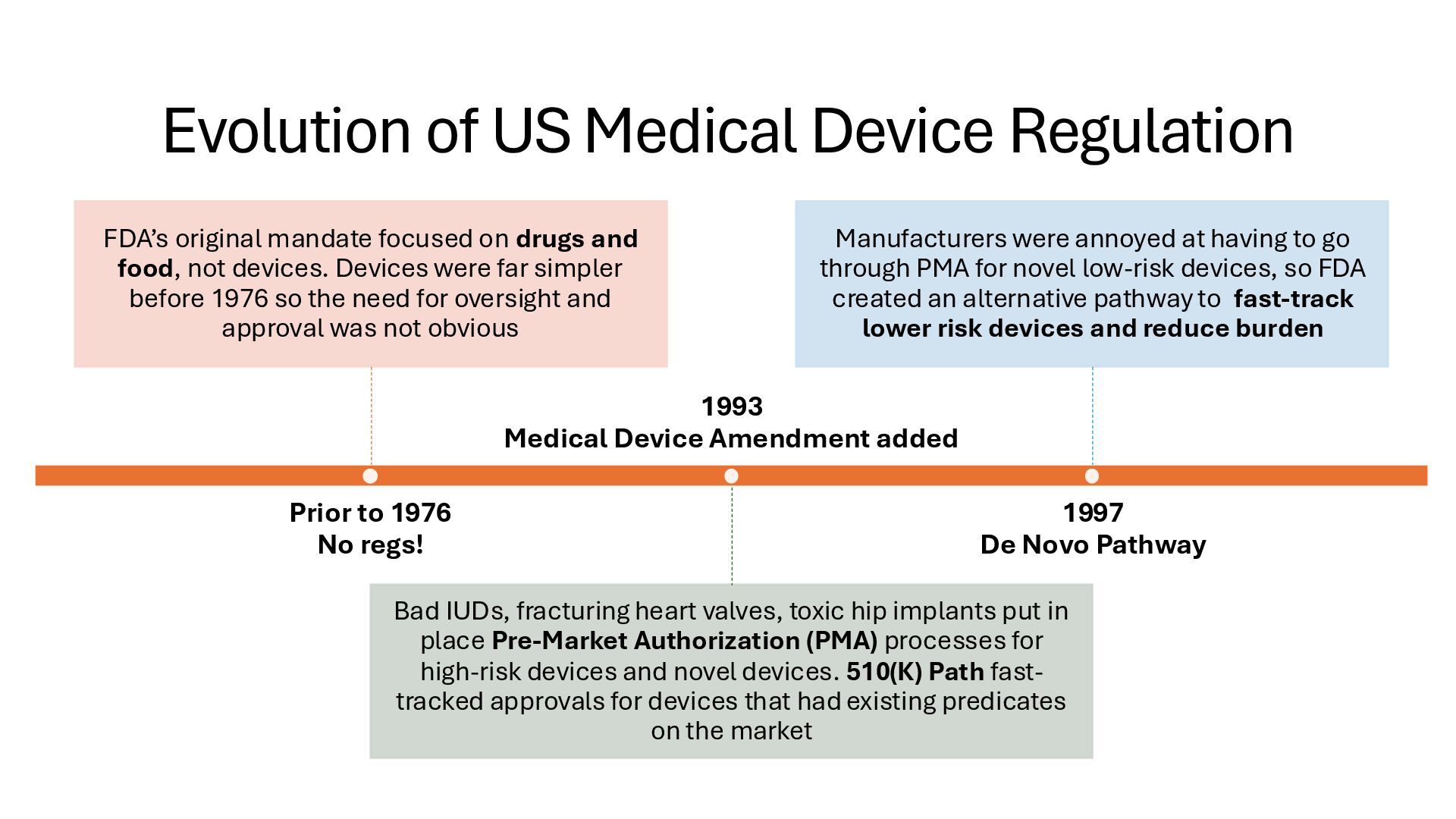
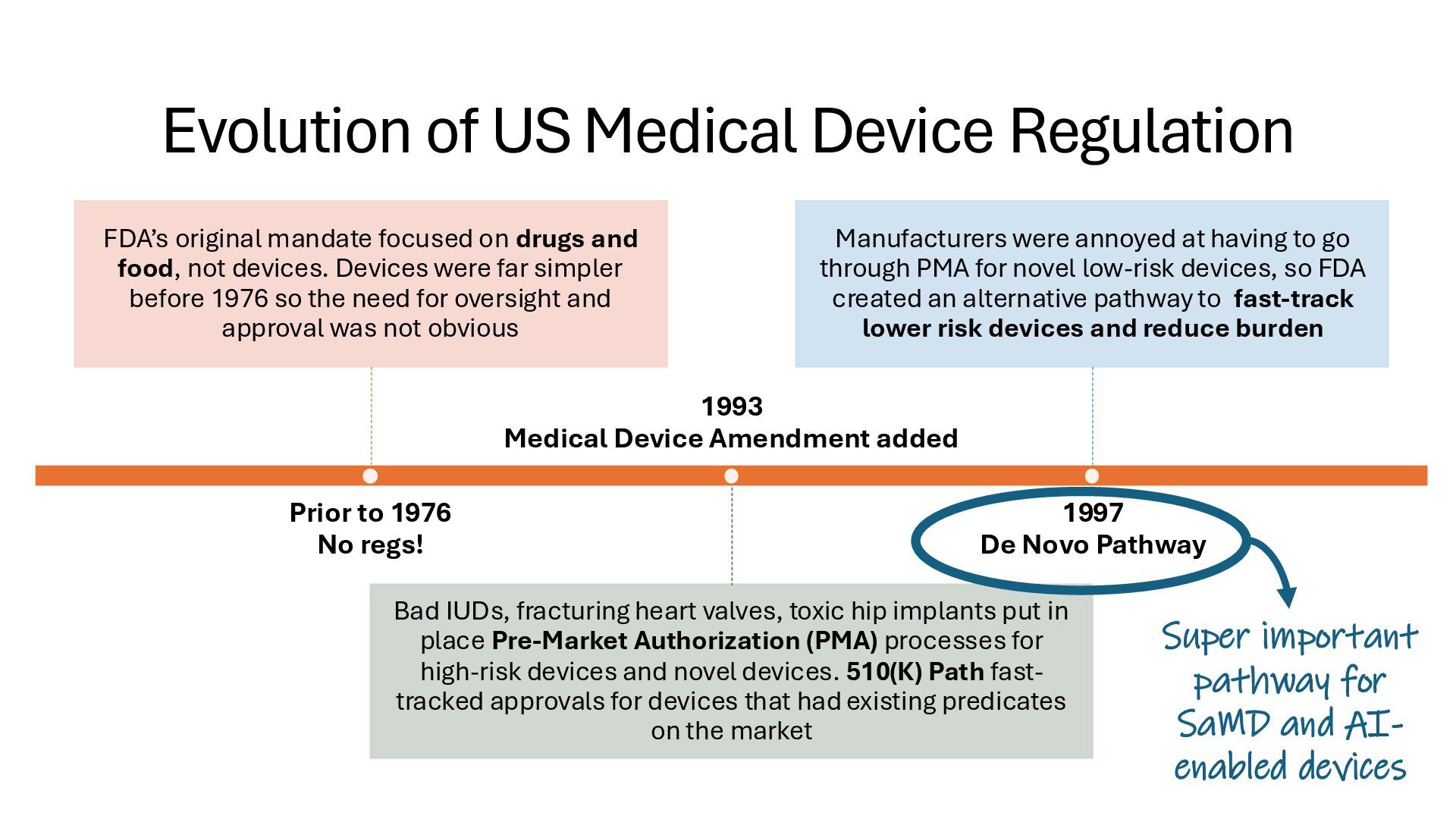
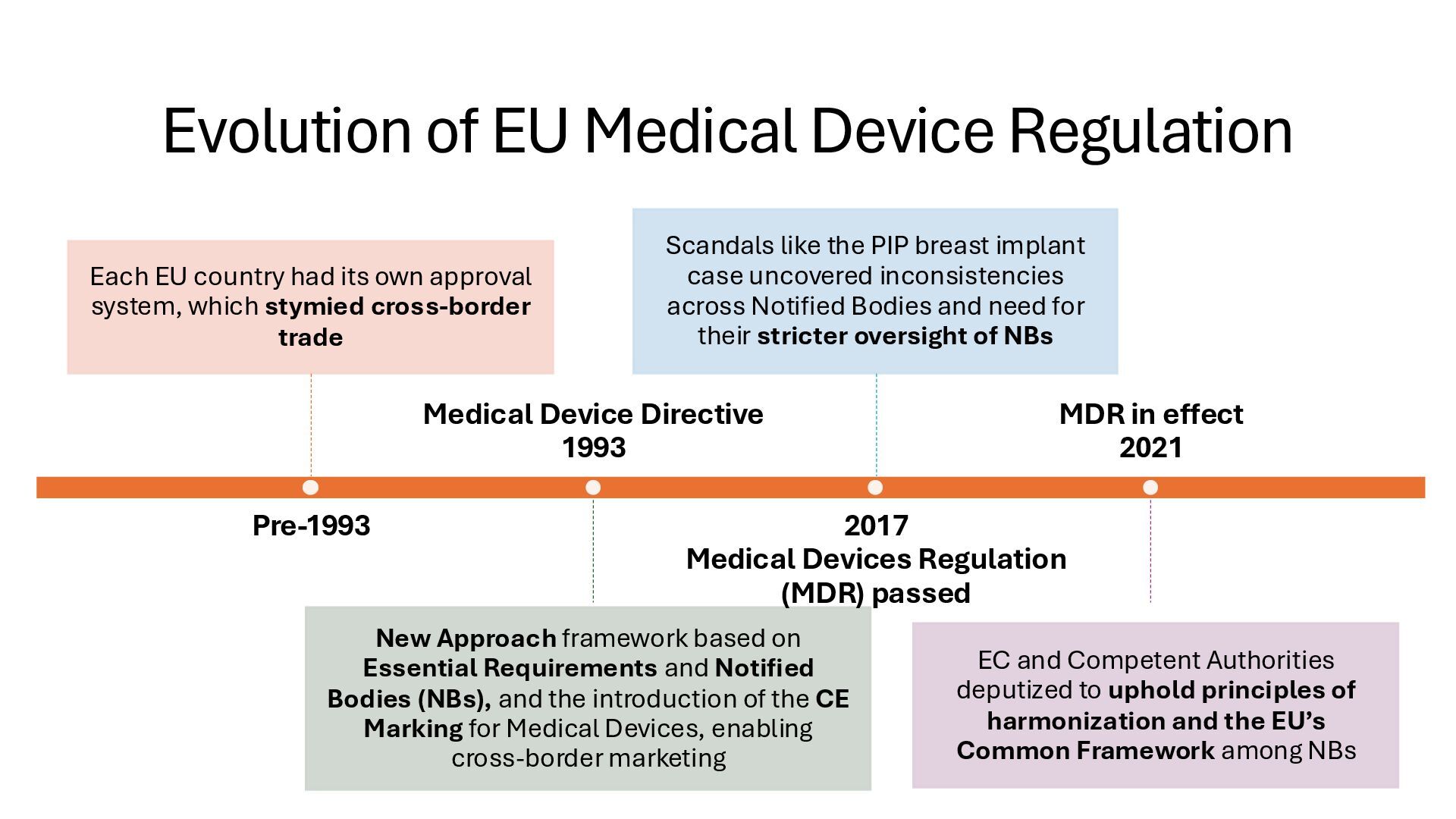
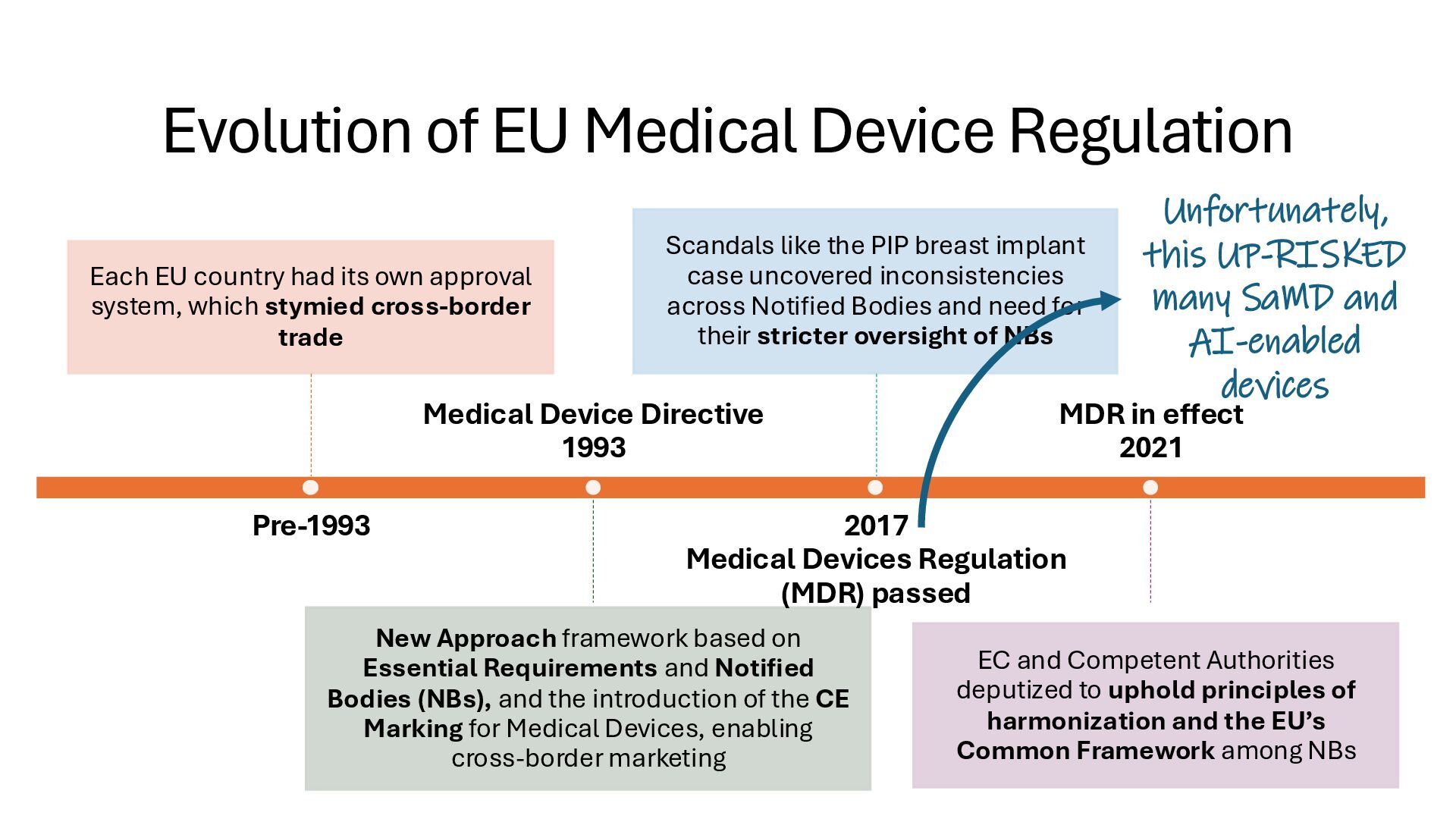
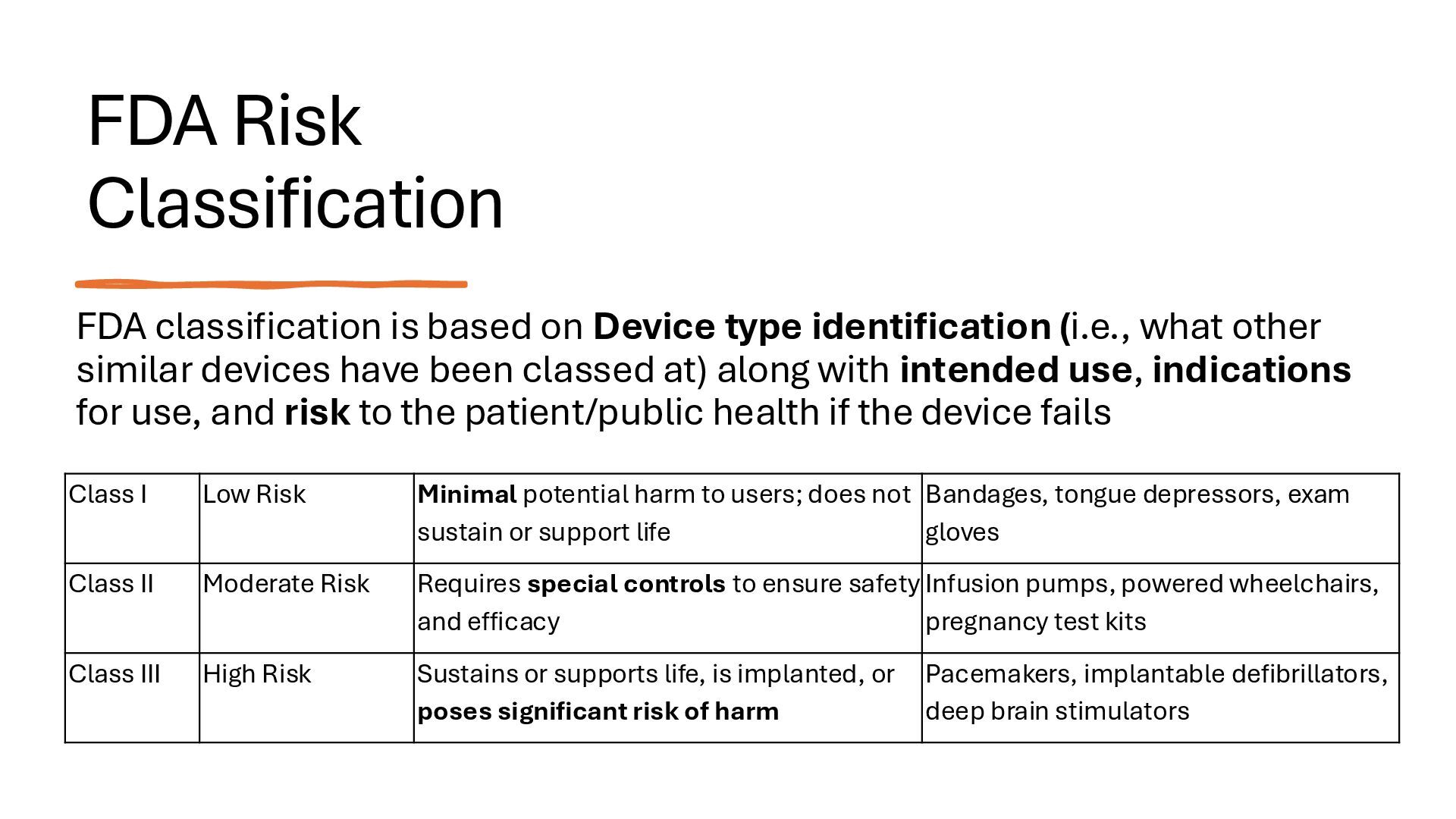
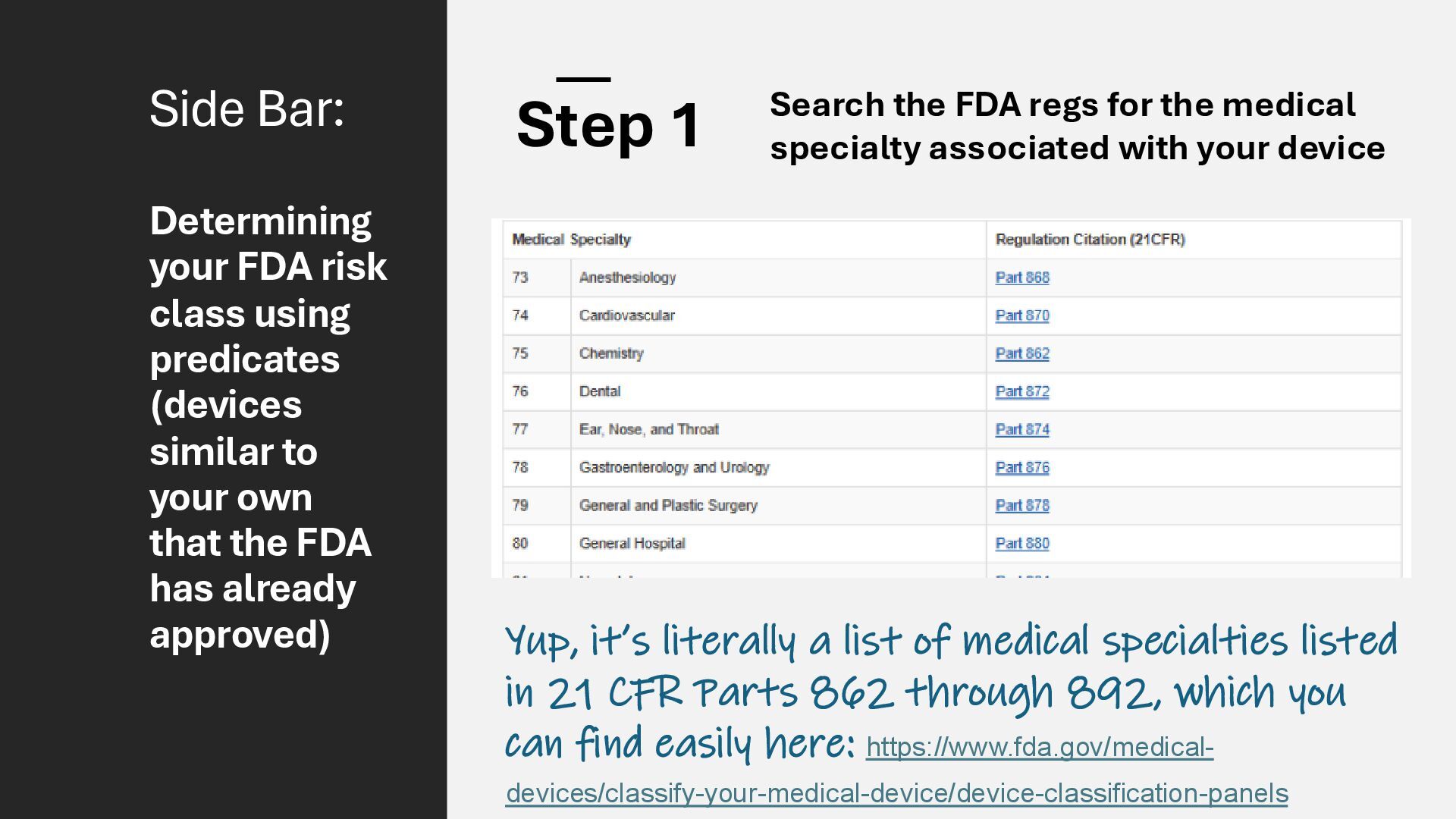
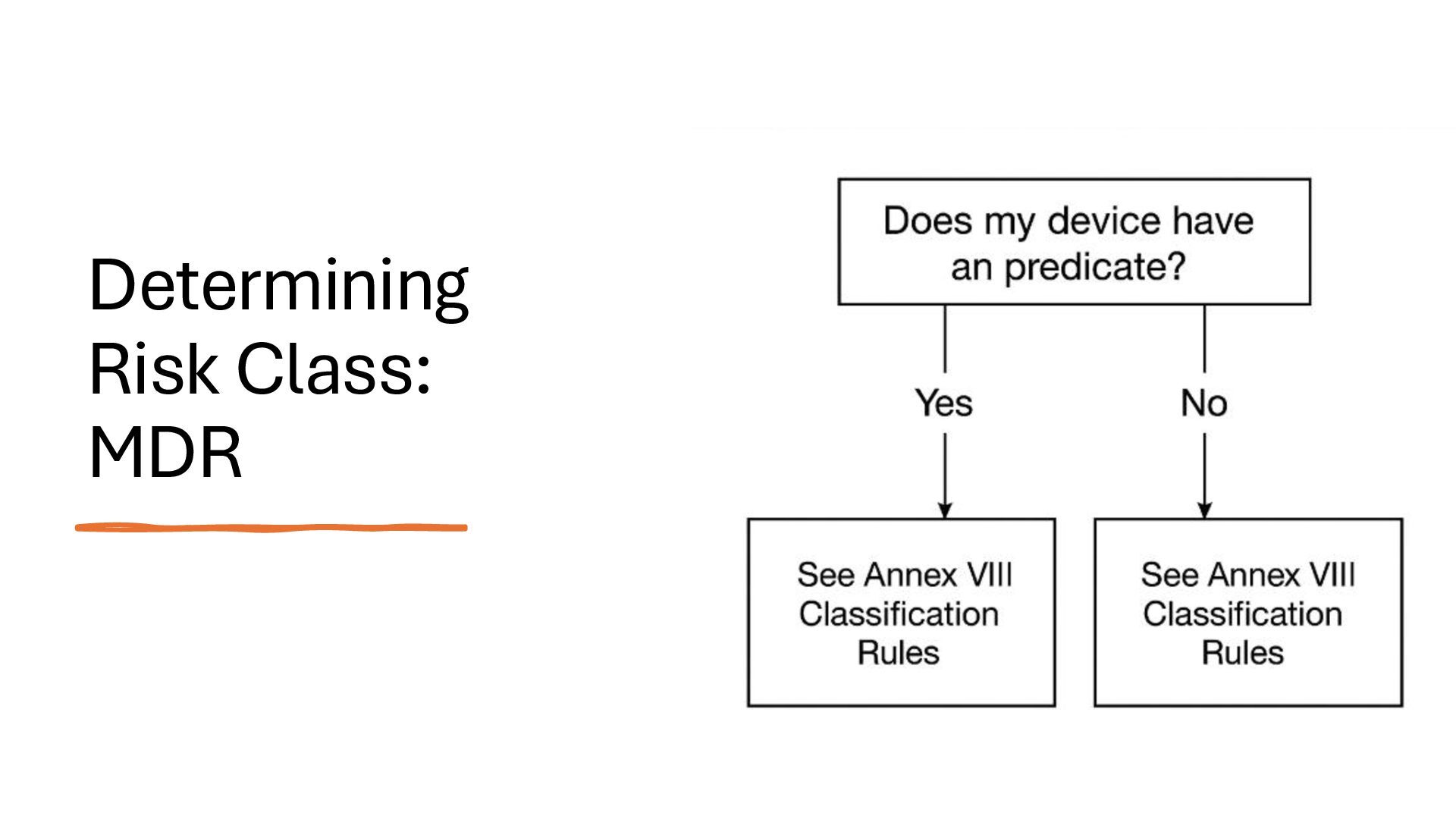
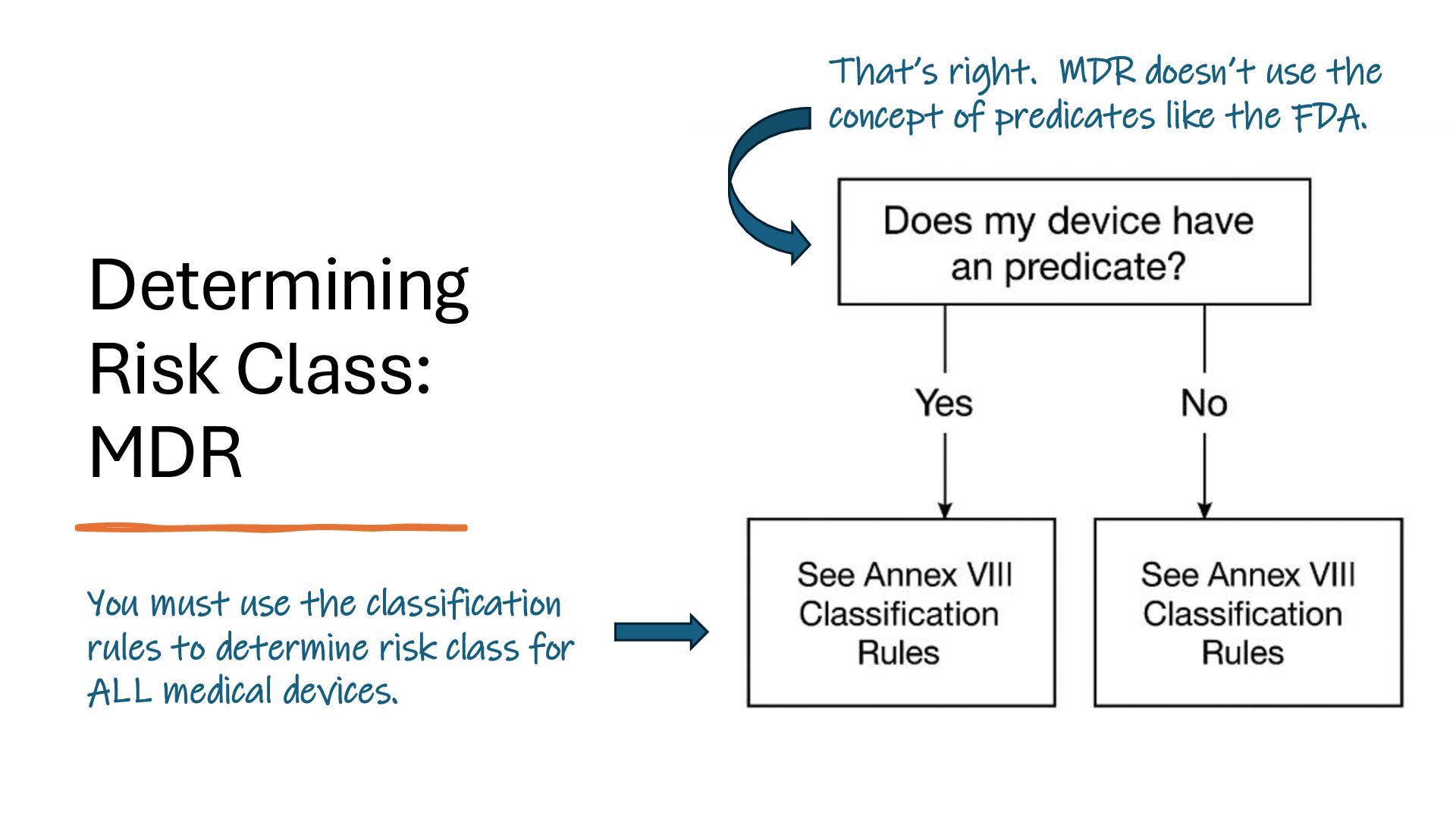
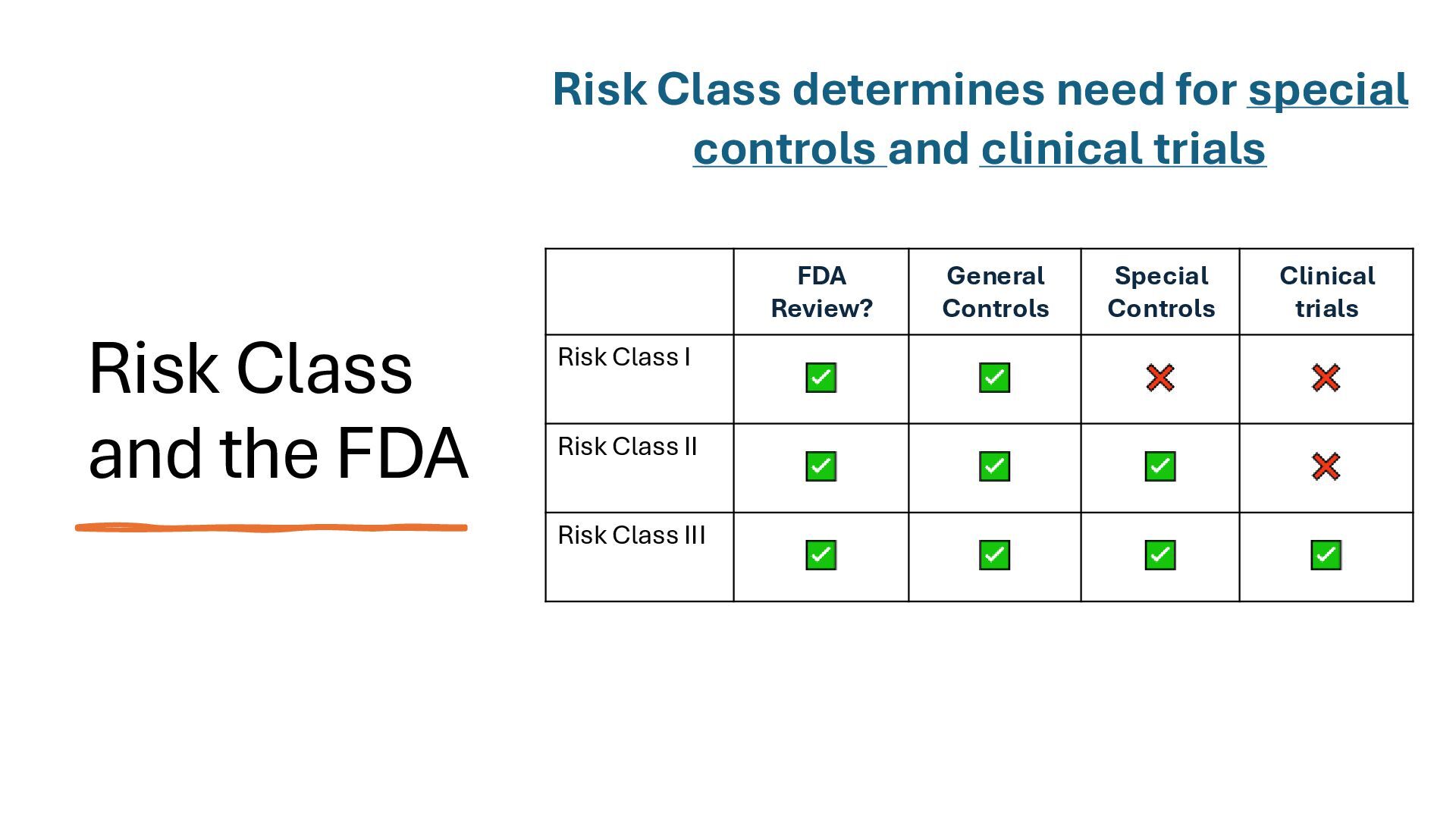
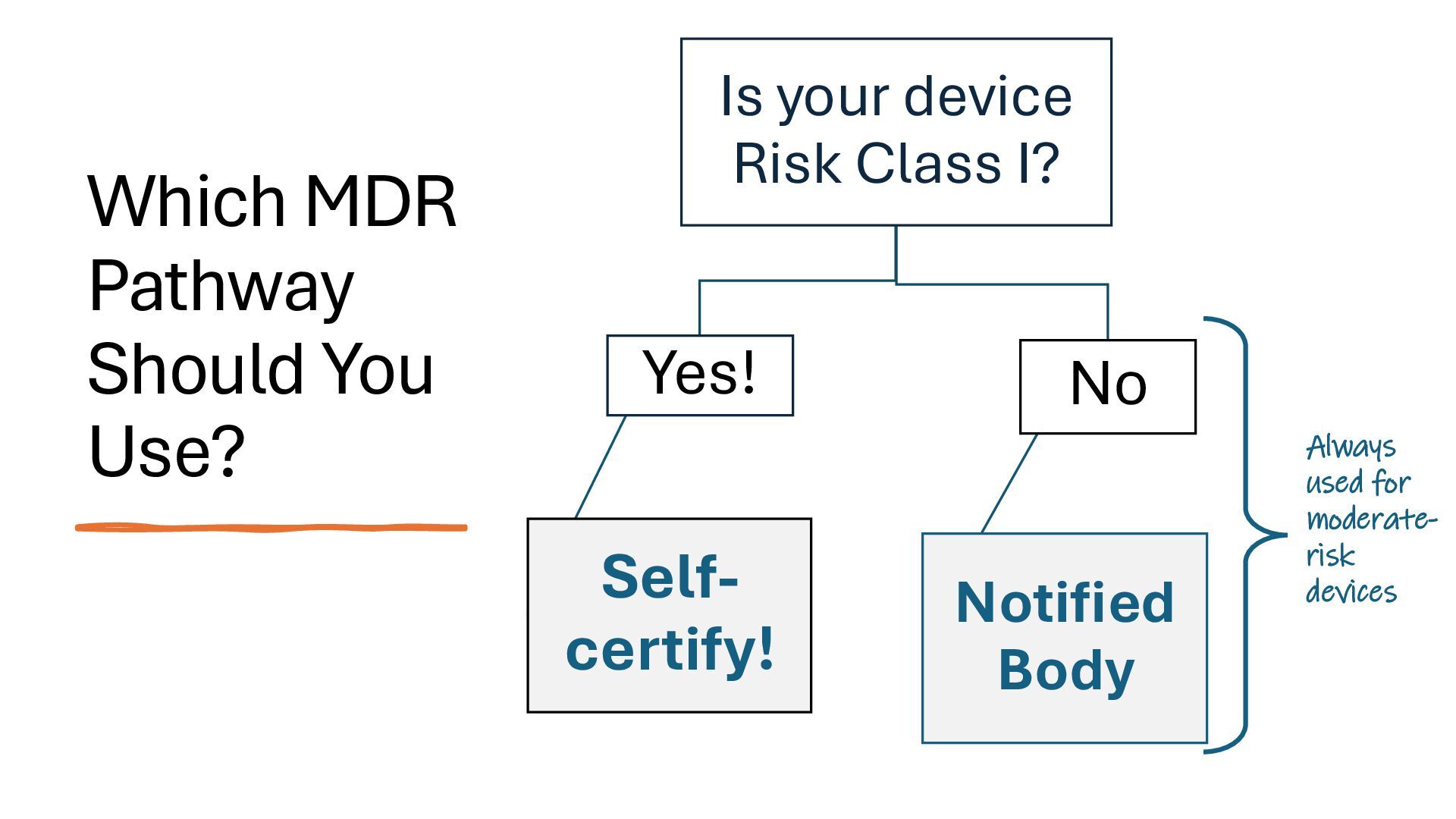
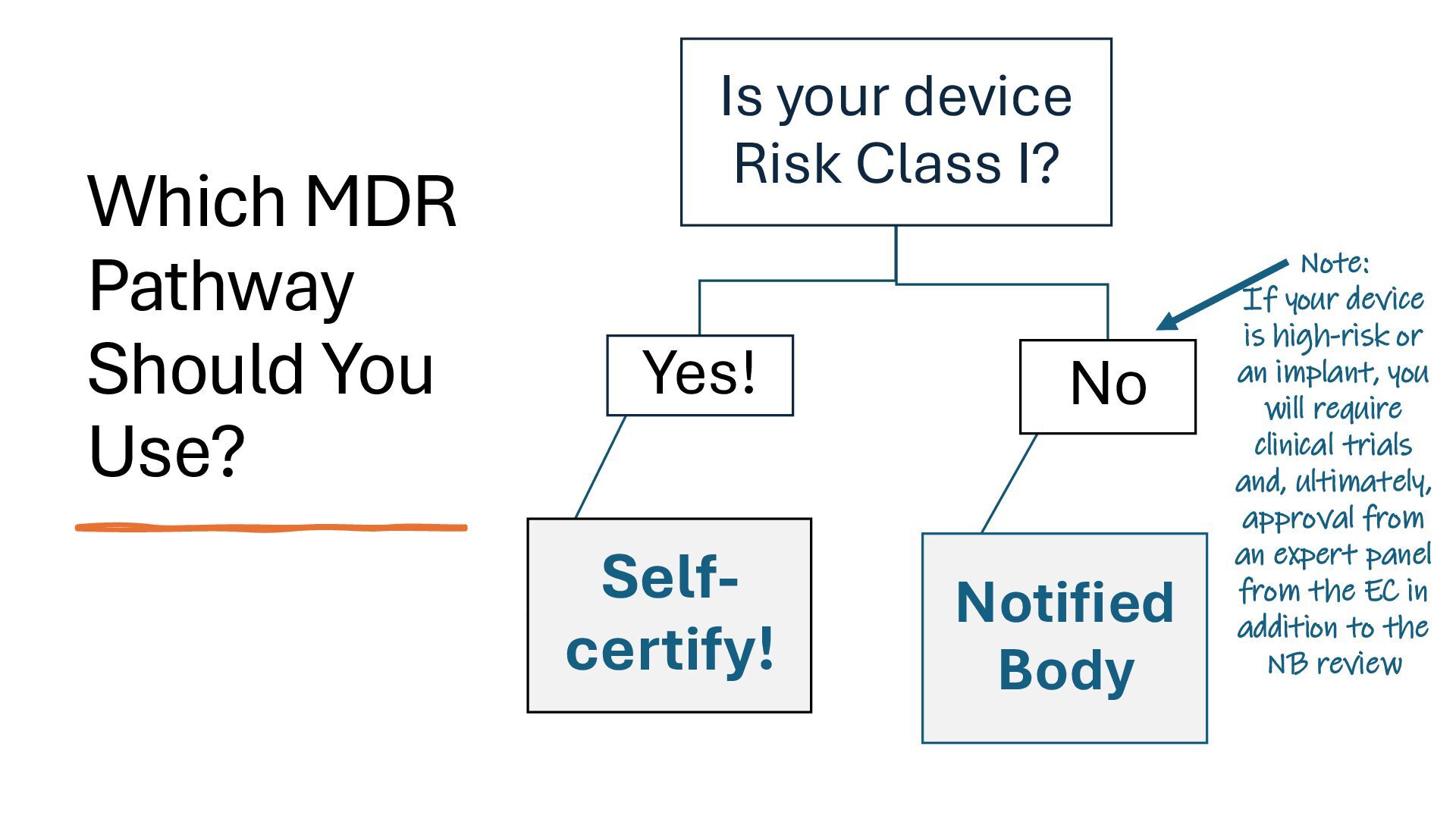
identification (i.e., what other similar devices have been classed at) along with intended use, indications for use, and risk to the patient/public health if the device fails Class I Low Risk Minimal potential harm to users; does not sustain or support life Bandages, tongue depressors, exam gloves Class II Moderate Risk Requires special controls to ensure safety and efficacy Infusion pumps, powered wheelchairs, pregnancy test kits Class III High Risk Sustains or supports life, is implanted, or poses significant risk of harm Pacemakers, implantable defibrillators, deep brain stimulators
{kind=link}
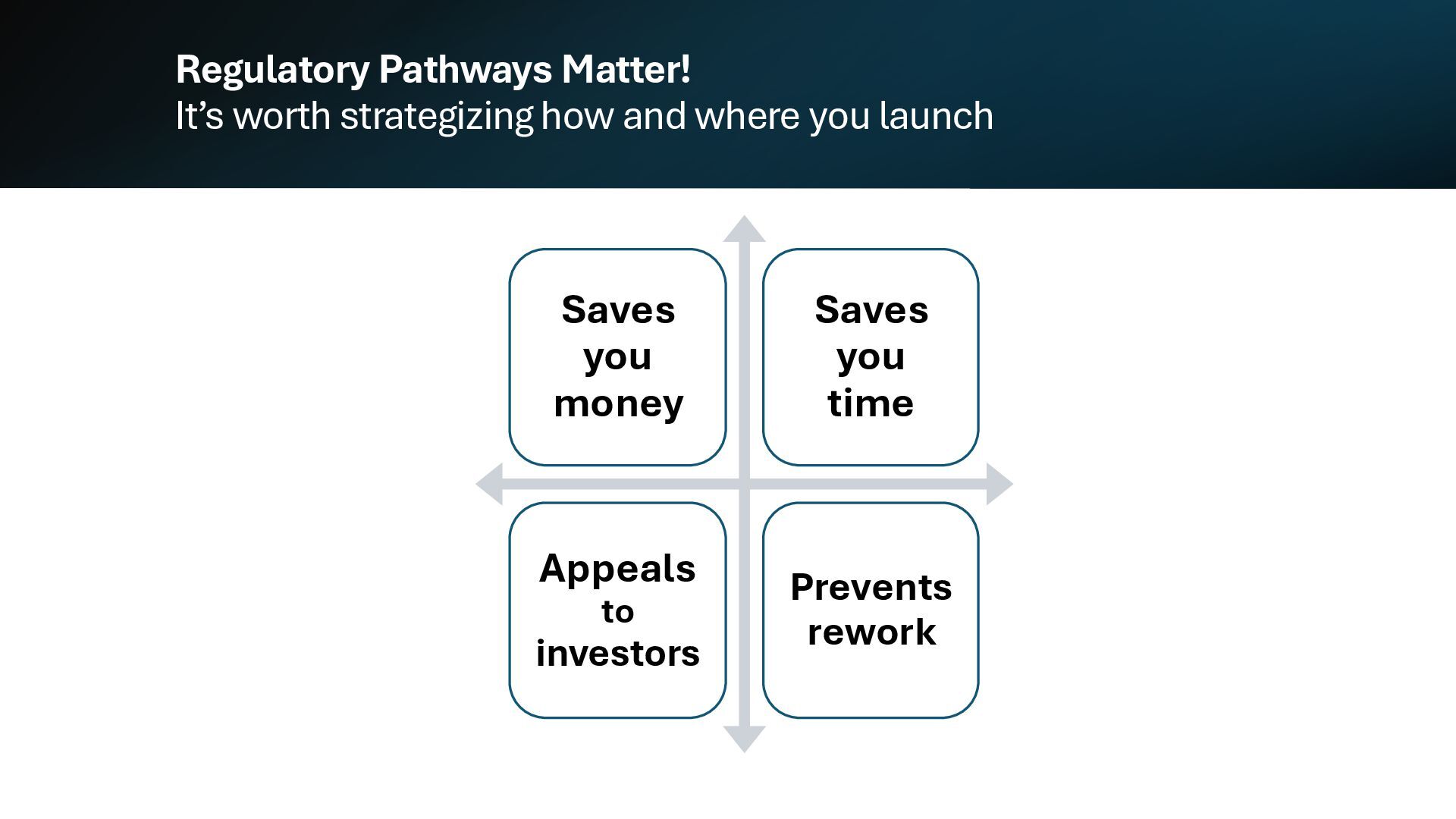{kind=link}
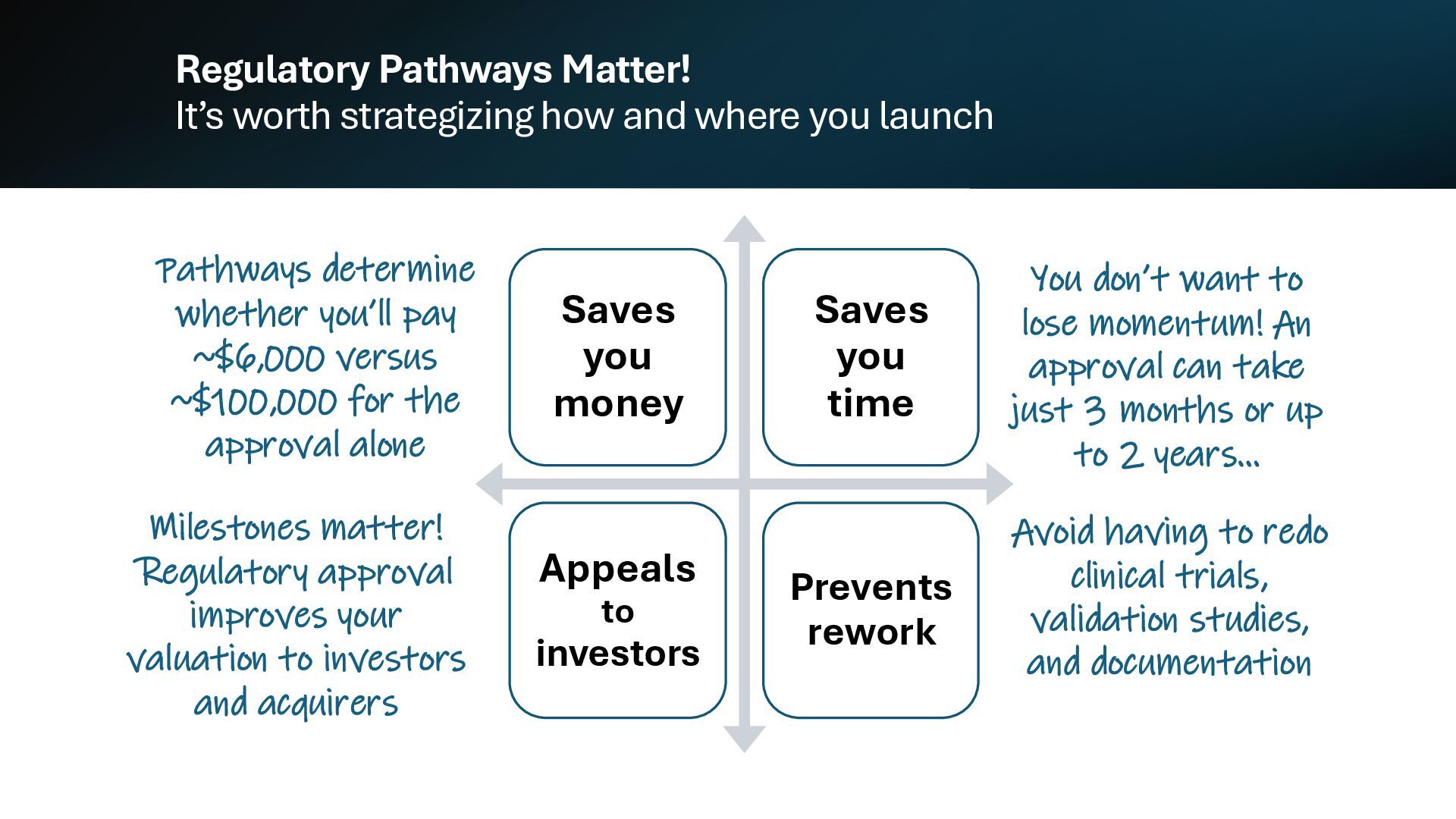{kind=link}
{kind=link}
{kind=link}
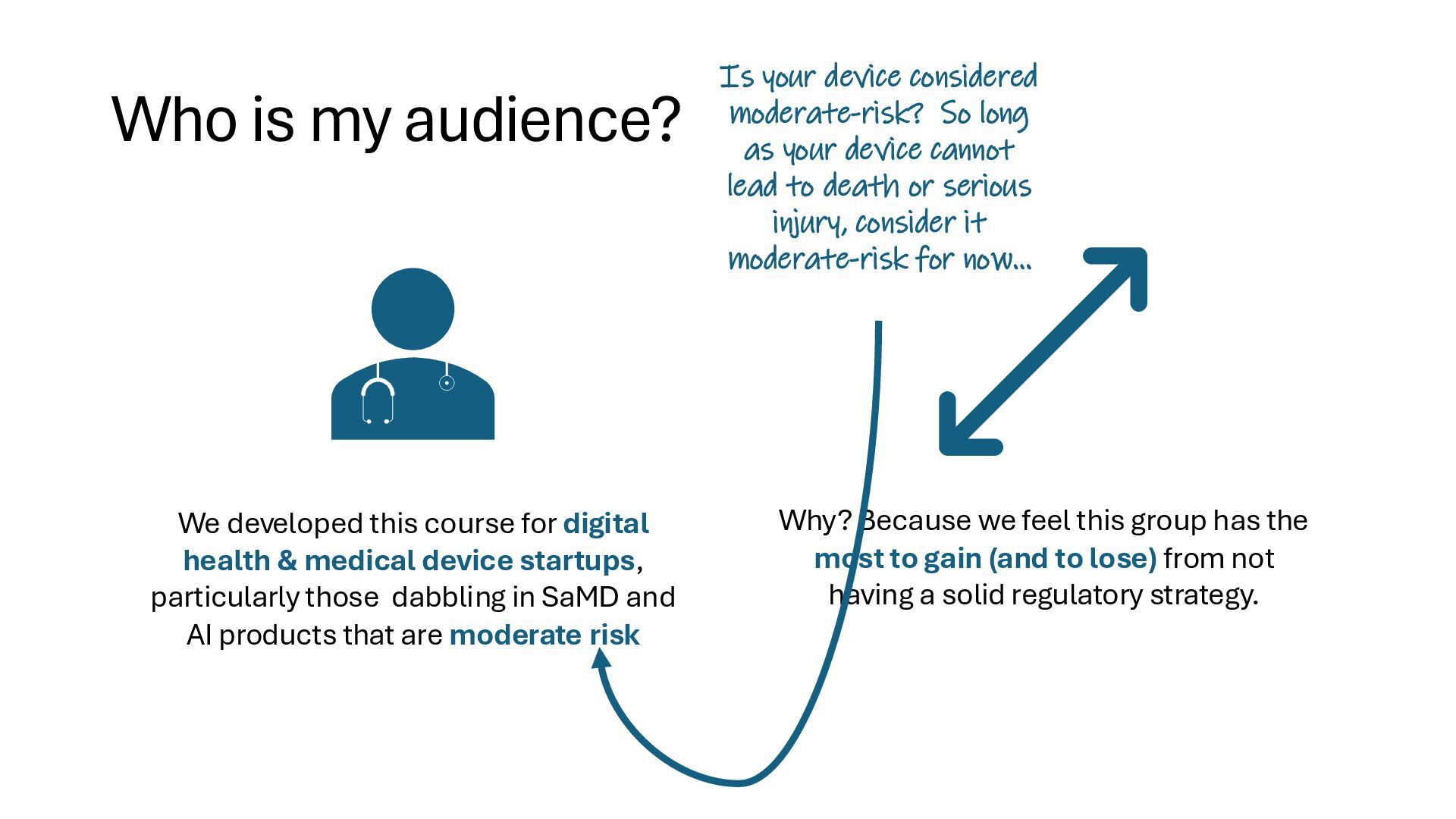{kind=link}
{kind=link}
{kind=link}
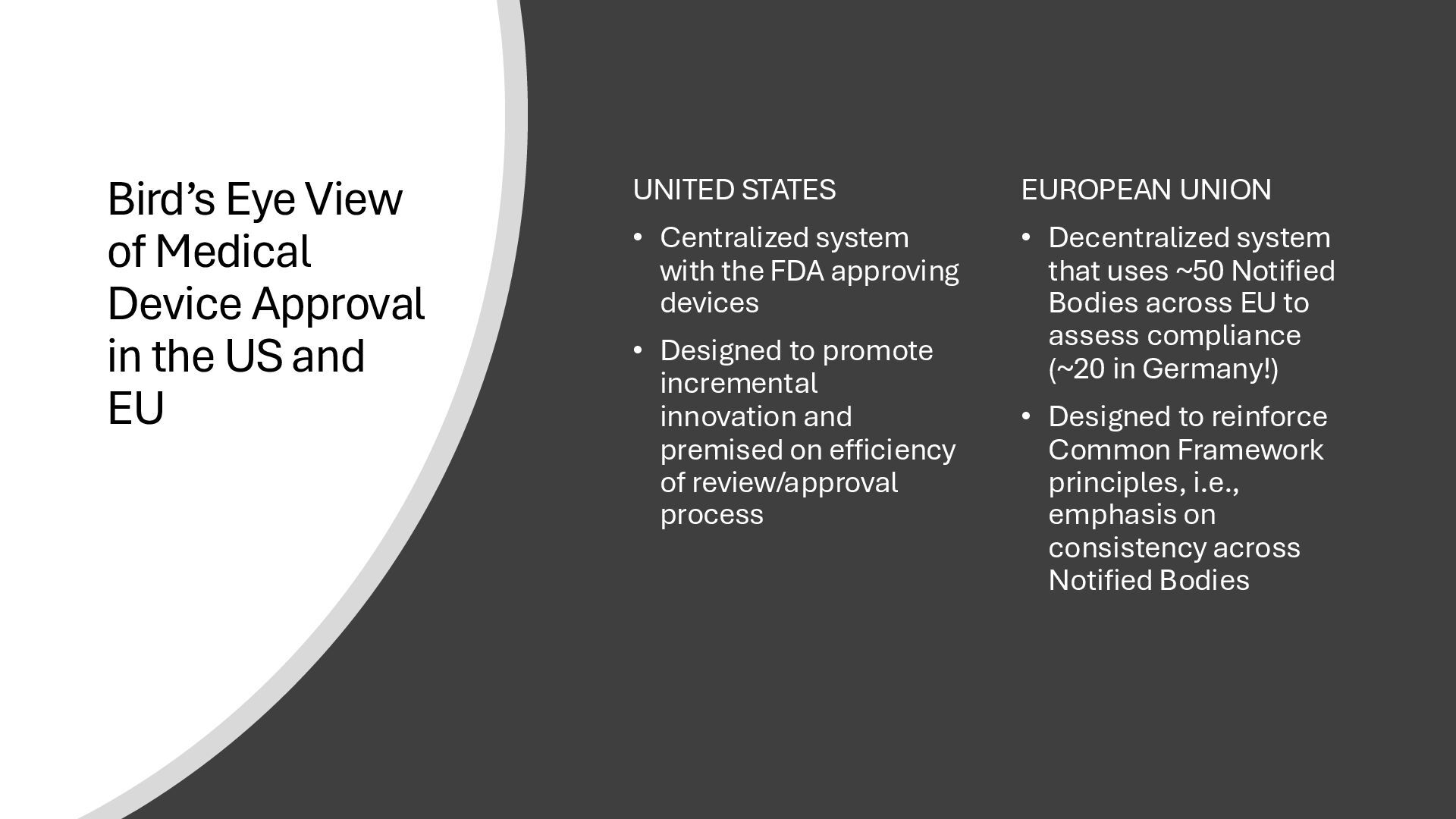{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}