6th International IBM Cloud Academy Conference 2018 25 May 2018 @ The Institute of Statistical Mathematics Tazro Ohta, Database Center for Life Science Tomoya Tanjo, National Institute of Informatics Osamu Ogasawara, National Institute of Genetics
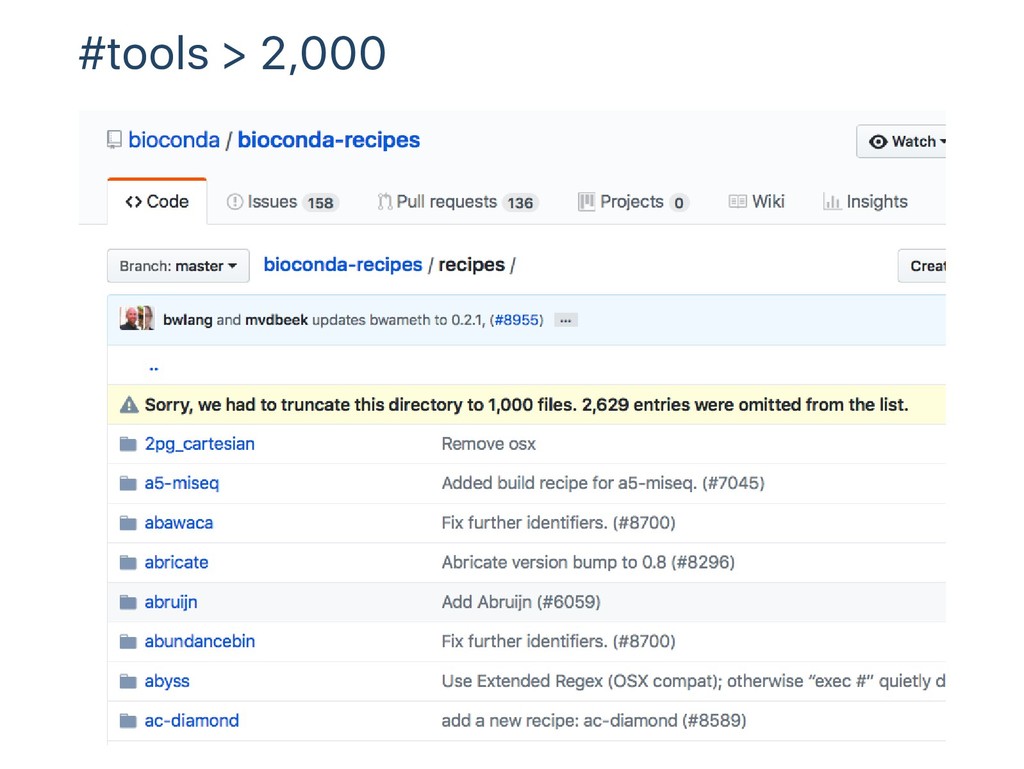
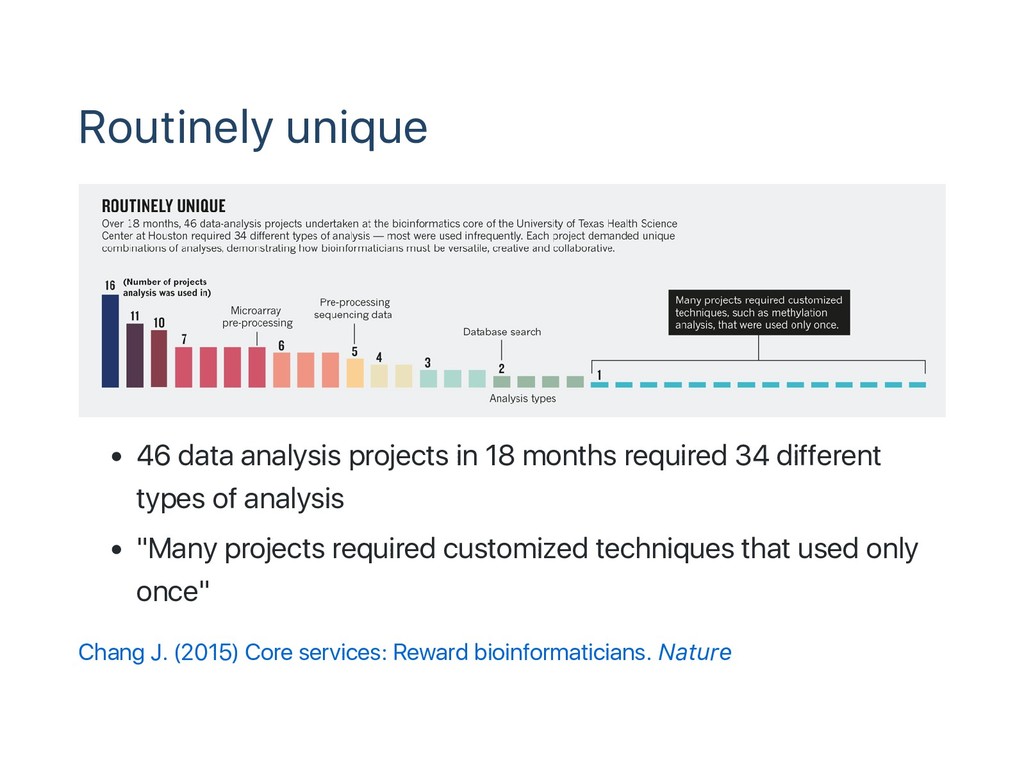
data analysis comes with Data explosion 100MB‑100GB per sample 10‑100,000 samples per research Large‑scale int'l collaborations / many individual projects Thousands of software tools Open source command line tools from individual developers "Workflow" to connect tools and iterate for samples "Routinely unique"
34 different types of analysis "Many projects required customized techniques that used only once" Chang J. (2015) Core services: Reward bioinformaticians. Nature
workflows Transfer data Fetch reference data from the public databases Manage preprocessing jobs Set up interactive environment for statistics (e.g. Jupyter) Collect and keep result data Repeat on reviewer's demand Can clouds help us to reduce the cost of the routines?
"the observed standard deviation is smaller when running with Docker" Di Tommaso P, et al. (2015) The impact of Docker containers on the performance of genomic pipelines. PeerJ
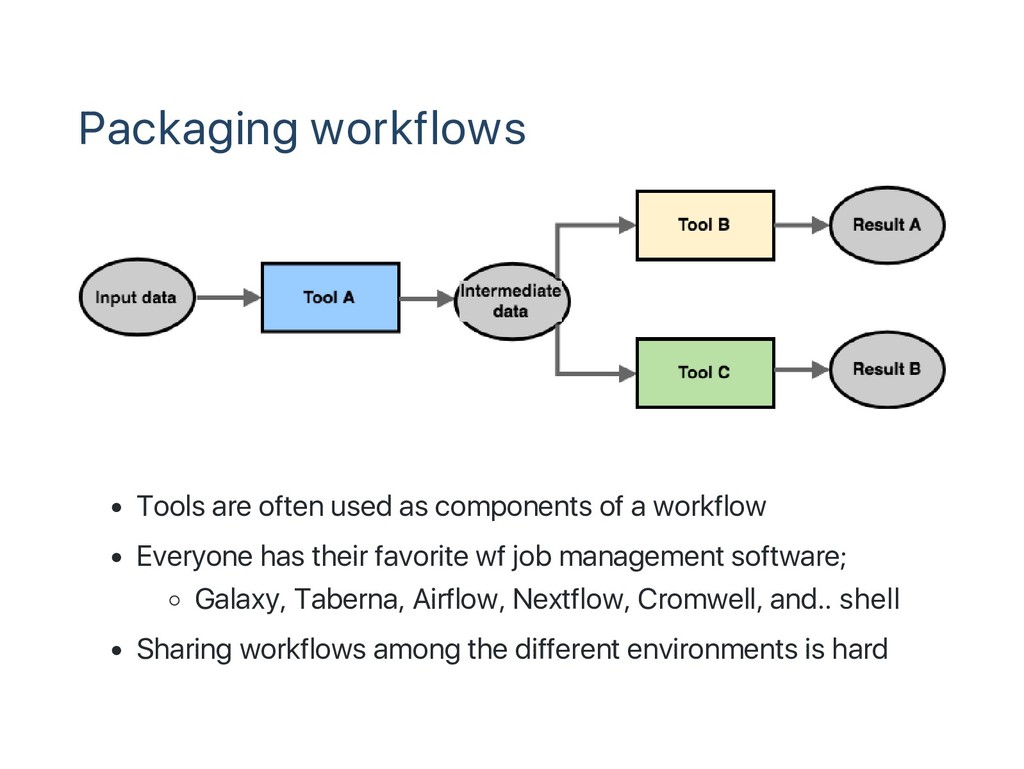
workflow Everyone has their favorite wf job management software; Galaxy, Taberna, Airflow, Nextflow, Cromwell, and.. shell Sharing workflows among the different environments is hard
analysis workflows and tools" A new open‑source community standard since 2014 for all data analysis tasks, not only bioinformatics describes structure of tools and workflows in YAML base container image, base command, input/output
e r s i o n : v 1 . 0 c l a s s : C o m m a n d L i n e T o o l h i n t s : D o c k e r R e q u i r e m e n t : d o c k e r P u l l : i n u t a n o / r s e m : 0 . 1 . 0 b a s e C o m m a n d : [ " r s e m - c a l c u l a t e - e x p r e s s i o n " ] i n p u t s : t h r e a d s : t y p e : i n t i n p u t B i n d i n g : p r e f i x : - p f a s t q : t y p e : F i l e i n p u t B i n d i n g : p o s i t i o n : 1 o u t p u t s : r e a d s P e r G e n e : t y p e : F i l e o u t p u t B i n d i n g : g l o b : " * R e a d s P e r G e n e . o u t . t a b "
e r s i o n : v 1 . 0 c l a s s : W o r k f l o w i n p u t s : r u n T h r e a d N : i n t d a t a U R L : s t r i n g o u t p u t s : r e a d s P e r G e n e : t y p e : F i l e o u t p u t S o u r c e : r s e m / r e a d s P e r G e n e s t e p s : d o w n l o a d _ d a t a : r u n : d o w n l o a d _ d a t a . c w l i n : d a t a U R L : d a t a U R L o u t : [ d a t a F i l e s ] r s e m : r u n : r s e m . c w l i n : t h r e a d s : r u n T h r e a d N d a t a : d o w n l o a d / d a t a F i l e s o u t : [ r e a d s P e r G e n e ]
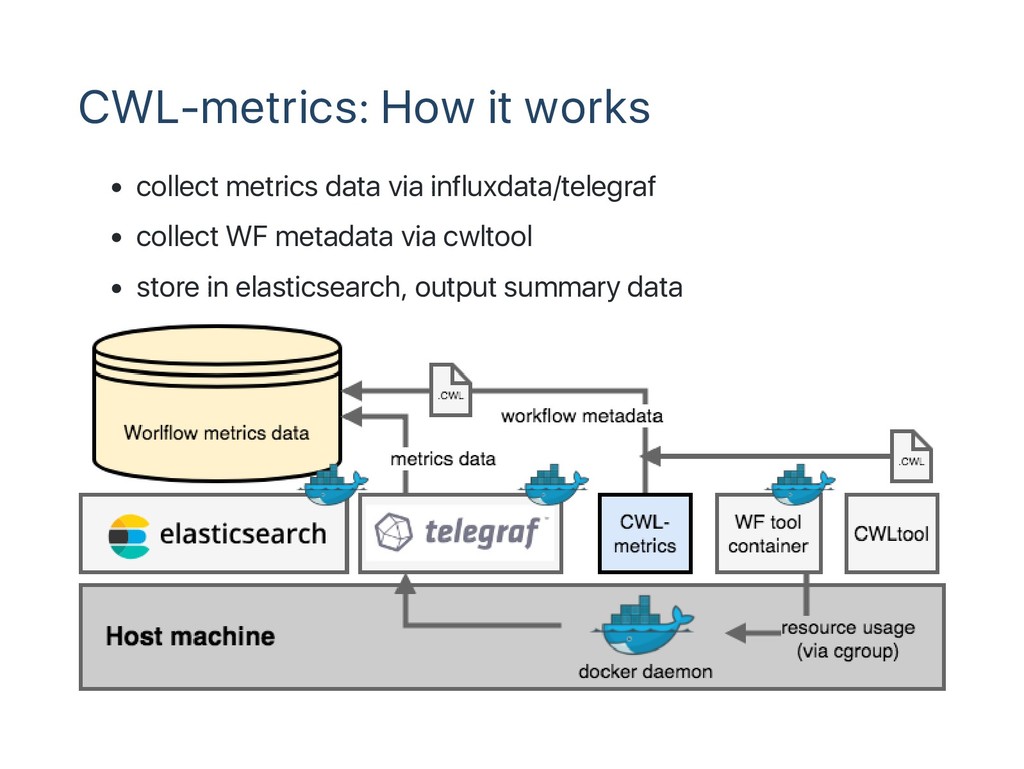
$ c w l t o o l r s e m _ w o r k f l o w . c w l r s e m _ w o r k f l o w _ j o b c o n f . y m l Basic idea CWL focuses on "What" of workflow structure of workflow incudling input, action, and output "How" should be determined by execution environment job scheduling and management are depending on engines
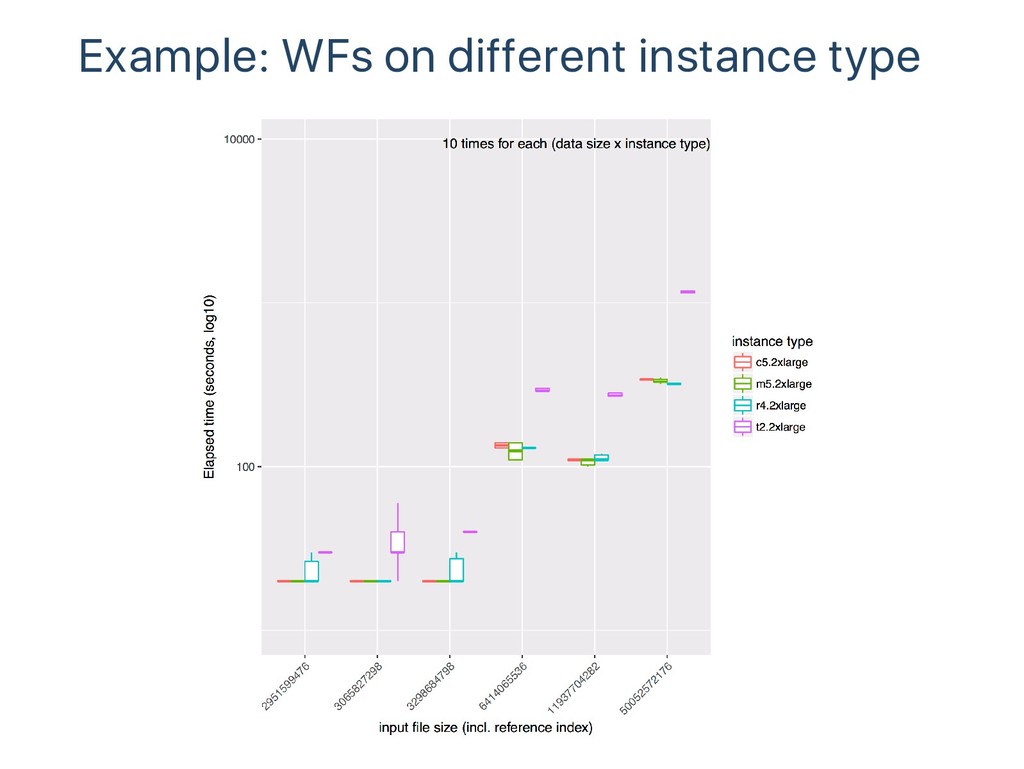
file Support more workflow engines Support multihost environment Support containers other than docker Summary Genomics need more machines, more easy‑to‑use clouds Packaging tools and workflows for easy migration to the clouds Collecting data for environment selection optimization Need more metrics data of wider variety of workflows
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}