Upgrade to Pro
— share decks privately, control downloads, hide ads and more …
Speaker Deck
Features
Speaker Deck
PRO
Sign in
Sign up for free
Search
Search
oku-slide-20240802
Search
Makito Oku
July 30, 2024
Research
0
97
oku-slide-20240802
幾つかのデータ解析手法の紹介
奥 牧人
2024/08/02
富山大学ムーンショット研究数理セミナー
Makito Oku
July 30, 2024
Tweet
Share
More Decks by Makito Oku
See All by Makito Oku
oku-slide-20231129
okumakito
0
120
oku-slide-20230827
okumakito
0
130
oku-slide-20230213
okumakito
0
230
oku-slide-20221212
okumakito
0
87
oku-slide-20221129
okumakito
0
160
oku-slide-20221115
okumakito
0
310
oku-slide-20220820
okumakito
0
330
oku-slide-stat1-1
okumakito
0
270
oku-slide-stat1-2
okumakito
0
300
Other Decks in Research
See All in Research
Weekly AI Agents News! 12月号 論文のアーカイブ
masatoto
0
250
Vision Language Modelと完全自動運転AIの最新動向
tsubasashi
1
310
言語モデルの内部機序:解析と解釈
eumesy
PRO
32
13k
Weekly AI Agents News! 1月号 アーカイブ
masatoto
1
220
20241226_くまもと公共交通新時代シンポジウム
trafficbrain
0
480
Prithvi-EO-2.0: A Versatile Multi-Temporal Foundation Model for Earth Observation Applications
satai
3
210
CoRL2024サーベイ
rpc
2
1.8k
JSAI NeurIPS 2024 参加報告会(AI アライメント)
akifumi_wachi
5
930
ノンパラメトリック分布表現を用いた位置尤度場周辺化によるRTK-GNSSの整数アンビギュイティ推定
aoki_nosse
0
220
[輪講] Transformer Layers as Painters
nk35jk
4
720
ラムダ計算の拡張に基づく 音楽プログラミング言語mimium とそのVMの実装
tomoyanonymous
0
440
請求書仕分け自動化での物体検知モデル活用 / Utilization of Object Detection Models in Automated Invoice Sorting
sansan_randd
0
140
Featured
See All Featured
ReactJS: Keep Simple. Everything can be a component!
pedronauck
666
120k
Learning to Love Humans: Emotional Interface Design
aarron
273
40k
Raft: Consensus for Rubyists
vanstee
137
6.8k
I Don’t Have Time: Getting Over the Fear to Launch Your Podcast
jcasabona
32
2.2k
CSS Pre-Processors: Stylus, Less & Sass
bermonpainter
356
30k
Designing for Performance
lara
606
69k
Practical Tips for Bootstrapping Information Extraction Pipelines
honnibal
PRO
16
1.1k
The Cult of Friendly URLs
andyhume
78
6.3k
How to Ace a Technical Interview
jacobian
276
23k
Typedesign – Prime Four
hannesfritz
41
2.6k
Distributed Sagas: A Protocol for Coordinating Microservices
caitiem20
331
21k
jQuery: Nuts, Bolts and Bling
dougneiner
63
7.7k
Transcript
幾つかのデータ解析手法の紹介 奥 牧人 2024/08/02 富山大学ムーンショット研究数理セミナー 発表後に一部修正 1 / 60
Outline はじめに 一般的な手法 同期性揺らぎ遺伝子解析 (DNB解析) エネルギー地形解析 RNA-seqデータ前処理 まとめ 2 /
60
Outline はじめに 一般的な手法 同期性揺らぎ遺伝子解析 (DNB解析) エネルギー地形解析 RNA-seqデータ前処理 まとめ 3 /
60
背景 富山大学ムーンショット研究のメンバーの多くは実験系であり、 データ解析を担当出来る 数理系の者が慢性的に不足 している。 特に、習得難易度の高いデータ解析手法の場合、手法毎に扱える 者が一人しかいない状況も珍しくない。 また、比較的簡単な解析でさえも一部の者に集中しがちである。 しかし、数理系の者を増やすのは難しい。 4
/ 60
目的 一人しか扱えない手法について、万一に備えて、せめて概要だけ でも 複数名で理解しておく ことを目指す。 比較的簡単な解析が出来る者 を増やす。 → まずはお互いに知っている手法を紹介 5
/ 60
その他の問題 (医薬系) 医薬系の場合 授業でプログラミングや機械学習をほとんど習わない。 稀にデータ解析の出来る学生や研究員が出現するが、卒業や 異動の際に知識やノウハウが誰にも継承されず失われる。 数理の課題について相談できる者が少ない。 データ解析主体では論文になりにくい。 対策 マニュアルや相談体制を整備し、研究が軌道に乗る最小人数
(critical mass) を越えることを目指す。 基本的に 実験系の者が自分でデータ解析 できるようにし、 数理の専門家は助言や指導に徹する状況を目指す。 6 / 60
その他の問題 (理工系) 理工系の場合 生命科学分野の実データが入手しづらい。 異分野のデータの場合、結果の解釈がしづらい。 標準的な手法を使っただけでは研究と認めて貰えない。 対策 公開データや人工データを使う。 良き共同研究相手 を見つける。ダメな相手は切る。
独自性の高い手法 を使いこなせるようにする。 7 / 60
以前まとめた資料 遺伝子発現量データ解析の基礎, 和漢研年報, 44:2-11 (2018). http://hdl.handle.net/10110/00019294 前処理、次元削減、発現変動遺伝子、ベン図、階層的クラス タリング、エンリッチメント解析、ネットワーク図を説明 上記解説文を元にした講義資料 https://speakerdeck.com/okumakito/oku-slide-20210721
動的ネットワークバイオマーカー解析の説明資料 https://speakerdeck.com/okumakito/oku-slide-20220820 8 / 60
何について学ぶべきか データ解析手法について学ぶとき、必ずしも 動作原理 や背後に ある 数学の概念 を理解する必要はない。 むしろ、実際のデータ解析で使用する際の 注意点 や、手法毎の
利点と欠点を踏まえた 使い分け を覚える方が大事。 自動車で例えると、エンジンの仕組みを詳細に理解していなくて も運転は出来るが、交通ルールを分かっていないと事故を起こし てしまう。 9 / 60
Outline はじめに 一般的な手法 同期性揺らぎ遺伝子解析 (DNB解析) エネルギー地形解析 RNA-seqデータ前処理 まとめ 10 /
60
統計学 医薬系では統計学をよく使う。医薬系の者の方が理工系の者より むしろ統計学に詳しい印象がある。 特に t検定 を多用する。 色々と批判されているものの、P値 を重視する者は多い。 私はそこまで詳しくないので、本発表では深入りしない。 11
/ 60
機械学習の区分のおさらい 大区分 中区分 例 教師あり学習 分類 線形判別分析、ロジスティック 回帰、ランダムフォレスト、 LightGBM 教師あり学習
回帰 単回帰、重回帰、Lasso, ガウス 過程回帰 教師なし学習 クラスタリング K平均法、階層的クラスタリング 教師なし学習 次元削減 PCA, t-SNE, UMAP 本発表では 教師なし学習のみ 扱う。 12 / 60
機械学習の区分の説明 教師あり学習は正解データあり、未知データの予測に使う。 教師なし学習は正解データなし、既存データの解釈に使う。 教師あり学習ではデータを訓練データとテストデータに分ける。 教師なし学習では全データをそのまま使う。 分類の出力は質的変数、回帰の出力は量的変数 クラスタリングではデータを複数のグループに分ける。 次元削減ではデータ点を (通常は) 2次元平面上に並べる。
ロジスティック回帰は、回帰と付いているが分類器 t-SNEやUMAPで通常グループ毎に色が違うのは、単に後から 追加でクラスタリングをかけているだけ 13 / 60
紹介する一般的な手法 主成分分析 t-SNE UMAP 倍率変化 FDR ベン図 階層的クラスタリング K平均法 エンリッチメント解析
14 / 60
主成分分析 項目 説明 名前 主成分分析、Principal Component Analysis (PCA) 区分 教師なし学習
> 次元削減 処理内容 多次元のデータを分散の意味で最もよく見える方向から 眺めた時の射影を計算する 関連用語 寄与率、ローディング、スクリープロット 利点 簡単、色々なことが分かる、解釈しやすい 欠点 scRNA-seqなど多数のグループがある場合に向かない 注意点 3次元は静止画だと意味がない (最適でない2次元への 射影になってしまっているため)、各軸毎に反転可 15 / 60
主成分分析、補足 私はPCAをデータ全体の傾向を把握する目的で使っている。 外れ値がないか、条件別に分かれているか、時間順に並んで いるかなどを確認 あくまで傾向把握目的なので、PC1, PC2のみ使用 重みの絶対値の大きい変数にも特に着目しない。重要変数の 選択はDEGでやる。 RNA-seqの場合、低発現遺伝子は予め除外する。 中心化
(平均を0にする) は必須だが、Zスコア化 (平均を0、標準 偏差を1にする) した方が良いかどうかはデータによって違う。 16 / 60
t-SNE 項目 説明 名前 t-SNE 区分 教師なし学習 > 次元削減 処理内容
多次元のデータ点を上手に2次元上に配置する 関連用語 - 利点 多数のグループをきちんと分離出来る 欠点 本当は同じグループの点が分離する場合がある、利用率 がUMAPに押され気味 注意点 sklearnの場合、学習率をデータ点数/12にし、初期配 置をPCAで決めるようにする。また、必要に応じて事前 にPCAで10次元程度に落としてからt-SNEをかける。 17 / 60
t-SNE、補足 t-SNEや後述のUMAPでは 各軸に優劣がない。寄与率 という概念 も存在しない。 簡単に言うと、 元の高次元空間で近い点は2次元平面上でもなるべく近く 元の高次元空間で遠い点は2次元平面上でもなるべく遠く なるように、各点の2次元平面上の位置をうまく調節する手法 つまり、PCAとは根本的に考え方が違う。
18 / 60
UMAP 項目 説明 名前 UMAP 区分 教師なし学習 > 次元削減 処理内容
多次元のデータ点を上手に2次元上に配置する 関連用語 - 利点 多数のグループをきちんと分離出来る 欠点 本当は同じグループの点が分離する場合がある、パラメ ータ調節が必要 注意点 パラメータ (min_dist, spread) を適宜調節する 19 / 60
倍率変化 項目 説明 名前 倍率変化、Fold change (FC) 区分 発現変動遺伝子解析 処理内容
2群間の比較において、平均値の比が (対数を取る前の 値で) 2倍より大きい遺伝子を選ぶ 関連用語 - 利点 簡単、再現性が高い、サンプル数に依存しない 欠点 P値至上主義者に理解して貰えない 注意点 閾値は2倍から変えても構わない 20 / 60
倍率変化、補足 複数時点ある場合、私は以下のようにしている。 各時点で平均を取る。 それらの最大値と最小値の差が (log2を取った値で) 1より 大きい遺伝子を選ぶ。任意の2時点間のDEGの和集合である。 取れた遺伝子を階層的クラスタリングにかける。 クラスタ毎に遺伝子一覧を調べたりエンリッチメント解析に かけたりする。
こうする理由は、多くの場合にDEGは複数時点で大きく重複する ので、時点別で見るよりクラスタ別で見た方が解釈しやすいから である。 21 / 60
FDR 項目 説明 名前 FDR、False Discovery Rate 区分 発現変動遺伝子解析、多重検定補正 処理内容
N個のP値に対応するN個のQ値を計算し、Q値が指定し たFDRの値以下のものを有意と判定する。FDRとは出力 の中に含まれる偽陽性の割合のこと。 関連用語 Benjamini-Hochberg法 利点 Nが大きい場合にBonferroni補正より検出力が高い 欠点 取れる遺伝子数がサンプル数に依存する 注意点 P値がほぼ同じでもQ値が大きく違ったり、P値が違うの にQ値が全く同じだったりする。P値とQ値は別物。 22 / 60
ベン図 項目 説明 名前 ベン図、Venn diagram 区分 発現変動遺伝子解析 処理内容 複数の集合間の重なり具合を計算して図示する
関連用語 和集合、積集合、差集合、ジャカード指数 利点 3集合以下なら分かりやすい 欠点 5集合以上だと領域数が多過ぎて意味不明 注意点 4集合の場合は正円だと描けないので楕円等を使う 23 / 60
階層的クラスタリング 項目 説明 名前 階層的クラスタリング 区分 教師なし学習 > クラスタリング 処理内容
似ているデータ点を順にまとめていき、閾値で切って 複数のクラスタに分ける 関連用語 樹形図、平均連結法、ウォード法、シルエットスコア 利点 ランダム性がない、生命科学の分野で一般的 欠点 汎用的な閾値自動決定法が無い 注意点 RNA-seqでは相関類似度を使うか遺伝子毎にZスコア化 する、単連結法はChaining現象が発生するため非推奨 24 / 60
階層的クラスタリング、補足 設定項目 類似度 or 非類似度: ユークリッド距離、相関類似度など 連結法: ウォード法、平均連結法など クラスタ分割基準: クラスタ数指定、非類似度の閾値指定など
私はRNA-seqデータ解析の場合では相関類似度、平均連結法、 非類似度の閾値指定を使っている。閾値は以下の式で計算 意味は、相関係数をフィッシャー変換した際の3σ点 1 − tanh ( 3 √N − 3 ) 25 / 60
K平均法 項目 説明 名前 K平均法、K-means 区分 教師なし学習 > クラスタリング 処理内容
クラスタ数をKとし、各クラスタの中心位置 (最初はラ ンダム) とそれに属するデータ点を交互に更新する 関連用語 - 利点 簡単 欠点 クラスタ数を決め打ちする必要がある、クラスタの初期 配置により結果が変わる 注意点 RNA-seqでは相関類似度を使うか遺伝子毎にZスコア化 する 26 / 60
エンリッチメント解析 項目 説明 名前 エンリッチメント解析 区分 その他 処理内容 入力した遺伝子一覧の中に、どのような注釈を持つ遺伝 子が多いかをデータベースに問い合わせ、出力する
関連用語 GO (Gene Ontology)、フィッシャーの正確検定 利点 個々の遺伝子について知らなくても結果の解釈が出来る 欠点 データベースによって結果が違う、重複数が極めて少な いものも出力される、詳しいことは分からない 注意点 P値だけ見てはダメ、個数も確認する 27 / 60
エンリッチメント解析、補足 フィッシャーの正確検定とは、ベン図で2つの集合の重なりが 有意に大きいかどうかを判定するもの。 エンリッチメント解析の場合の2つの集合とは、入力した遺伝子 群 (DEGなど) と特定の注釈を持つ遺伝子群のこと。 私は普段DAVIDを使うので、以下DAVIDについて補足する。 https://david.ncifcrf.gov/ P値はDAVIDの独自補正がかかっている。私はDAVIDから結果を
ダウンロードしてPythonでP値とQ値を再計算している。その 際、遺伝子の個数が少ない注釈も除外している。 GOで名前に"regulation of transcription" と付くものは解釈が 難しいので私は除外している。 28 / 60
Outline はじめに 一般的な手法 同期性揺らぎ遺伝子解析 (DNB解析) エネルギー地形解析 RNA-seqデータ前処理 まとめ 29 /
60
同期性揺らぎ遺伝子解析 (DNB解析) 項目 説明 名前 同期性揺らぎ遺伝子解析、DNB解析 区分 その他 処理内容 揺らぎが大きく、かつ、互いに強く相関した遺伝子群を
選ぶ 関連用語 中央絶対偏差、スピアマンの相関係数、階層的クラスタ リング 利点 揺らぎに注目する点が独自的 欠点 言葉が独り歩きしていて過度に期待される、結果が合っ ているかどうかよく分からない、手法が確立していない 注意点 後述 30 / 60
主な流れ 主な流れ 1. 揺らぎの大きな遺伝子を選ぶ 2. その中から相関強度の高いクラスタを選ぶ 必要に応じて前処理や後処理をする。 本質的に 変数選択 の手法、発現変動遺伝子と同様
31 / 60
考えるべきこと 考えるべきことが沢山ある。 揺らぎの指標: 標準偏差、中央絶対偏差 揺らぎ判定法: 倍率変化、割合、仮説検定 倍率変化や割合の閾値 仮説検定の種類: F検定、Levene ル
ビ ー ン 検定、Brown-Forsythe検定 対照群: 使用する、使用しない 複数時点の扱い: 時点毎、中心化して結合、対照群のみプール 相関の指標: ピアソンの相関係数、スピアマンの相関係数 連結法: 平均連結法、完全連結法 クラスタ分割基準: 個数、閾値 32 / 60
考えるべきこと、続き クラスタ選択: 最大クラスタのみ選択、2番目以降も選択 最大クラスタに対する大きさ 最大クラスタ数 最小クラスタサイズ 事後クラスタリング: する、しない 時点毎の中心化: する、しない
相関係数の指標: ピアソン、スピアマン 最大クラスタ数 最小クラスタサイズ 遺伝子全体の揺らぎスコア: 選択条件に使う、使わない 33 / 60
RNA-seqに対する現在の方針 外れ値に強い 中央絶対偏差 と スピアマンの相関係数 は必須 結果の再現性を高めるため 対照群なし を選択 時点毎に計算するか、中心化して結合するかは、結果があまり
変わらないことが多いのでどちらでも良い。 対照群なし、かつ、時点毎の場合、異なる時点間で取れる遺伝子 が大きく重複するので、事後クラスタリング が必要 結果の妥当性を以下で確認 ヒートマップ 遺伝子リスト エンリッチメント解析 34 / 60
何が難しいのか? 手法が確立しておらず、選択肢の組み合わせの数が膨大なため、 データ毎に 数理の知識 と データ解析の経験 に基づき組み合わせ を変え、その妥当性を高度な ドメイン知識 に基づき多角的かつ
慎重に判断する必要がある。 私自身もマイクロアレイとRNA-seqのドメイン知識しか持ってい ないので、他の種類のデータには全く手が出せない。 今回の目的はお互いに知っている手法を教え合うことだが、正直に言 うと DNB解析の習得は勧められない。やるとしても、再実験が容易 で結果の再現性を確認しやすいものが良いと思う。 35 / 60
よくある間違い 1サンプルのみで高発現し、残りのサンプルでは全然揺らいでい なかった → 外れ値 に引きずられただけ サンプルを番号順にした場合、あるサンプルまでは一様に低く、 それ以降が一様に高かった → バッチ効果
を拾っただけ snoRNA (Snorから始まる) など短い転写産物ばかり → S/N比 の悪い遺伝子が取れただけ Olfrなど平均発現量の低い遺伝子ばかり → 同上 ミトコンドリア遺伝子 (mtから始まる) やリボソーム蛋白質 (Rp から始まる) ばかり → 単に 高発現 遺伝子を拾っただけ ほぼ全ての条件で同じ遺伝子が出てきた → 単に コントロール群 で偶然値が揃っていた 遺伝子を拾っただけ 36 / 60
Outline はじめに 一般的な手法 同期性揺らぎ遺伝子解析 (DNB解析) エネルギー地形解析 RNA-seqデータ前処理 まとめ 37 /
60
エネルギー地形解析 項目 説明 名前 エネルギー地形解析、Energy Landscape Analysis (ELA) 区分 その他
処理内容 データを二値化し、イジングモデルに当てはめ、エネル ギーの谷や、谷と谷の間のエネルギー障壁を計算する 関連用語 イジングモデル、ベースングラフ、非連結性グラフ 利点 独自性が高い、解析ツールの使用自体は簡単 欠点 約7個の変数を選ぶ必要がある、結果の解釈が難しい 注意点 後述 38 / 60
エネルギー地形解析、補足 江崎先生がMatlabで作ったツールのPython移植版: https://github.com/okumakito/elapy エネルギーは仮想的なもので、確率と関連した値 単独のエネルギーの値に意味はなく、相対的な差が大事 頂点番号は、0と1から成る2進数を10進数に直したもの 例、1100111 = 64 +
32 + 4 + 2 + 1 = 103 ベースングラフでは、頂点が多いほど谷が広く、最小エネルギー が小さいほど谷が深いことを意味する。 非連結性グラフは、状態間 (正確にはエネルギー極小パターン間) を遷移する際のエネルギー障壁の高さを図示したもの。 p ∝ exp(−E) 2 39 / 60
エネルギー地形解析、補足続き 混乱を避けるため、ベースングラフの連結成分を 状態、個々の 頂点を パターン と呼び分ける。 ベースングラフの矢印は、1変数のみが異なる近傍パターンの中 でエネルギーが最小のものに向かって引いたもの、つまり、最急 降下方向を表す。実際は逆方向に遷移することもある。 非連結性グラフを元にした等高線図の作成も出来る。
40 / 60
Outline はじめに 一般的な手法 同期性揺らぎ遺伝子解析 (DNB解析) エネルギー地形解析 RNA-seqデータ前処理 まとめ 41 /
60
お断り ここから先はPythonのコードを載せる。 RNA-seqに興味の無い方にも、データフレームの使い方の参考に なるとありがたい。 データフレームとは、各行と各列の名前付きの2次元データ 42 / 60
主な前処理 FASTQ → BAM → カウントデータ (私は出来ないので外注) ファイルの結合 不要な行や列の除外 ID変換
列名の変更 正規化 対数化 外れ値の除外 低発現遺伝子の除外 その他、対象データ特有の問題に対する処理 43 / 60
ファイルの結合 データファイルはなるべく1つにまとめる。 作業効率を上げるため、かつ、ミスを減らすため 私の場合、結合したい元ファイルを同じディレクトリに入れ、 以下のような流れで結合している。後述の処理を適宜足す。 dir_name = '../data/orig/' # 元データを置いたディレクトリ名
file_list = os.listdir(dir_name) # ファイル名一覧を取得 file_list.sort() # 並び替え out_list = [] # 出力格納用リスト for file_name in file_list: file_path = os.path.join(dir_name, file_name) # フルパス df = pd.read_csv(file_path) # csvファイル読み込み ### ここに色々な処理を追加 ### out_list.append(df) # 出力格納用リストに追加 df = pd.concat(out_list, axis=1) # 全体を結合 df.to_csv('tmp.csv') # ファイルに保存、重要ファイル上書き防止のためtmp 44 / 60
ファイル読み込みの補足 データがコンマ区切りでなくタブ区切りの場合 df = pd.read_table(file_path) # read_csvの sep='\t' と同じ 1列目をindexとして扱う場合
(1行目は既定でheader扱い) df = pd.read_csv(file_path, index_col=0) 一部の列のみを読み込む場合 df = pd.read_csv(file_path, usecols=['ABC','DEF']) # 2列のみ ある文字から始まるコメント行を除く場合 df = pd.read_csv(file_path, comment='#') # '#" から始まる行を除外 他にもread_csv, read_tableのオプションは沢山ある。 45 / 60
不要な行や列の除外 ここは千差万別。比較的よく使うもの ある列 (仮にxyzとする) を除外 df = df.drop('xyz', axis=1) ある列が空欄でない行だけ抽出
df = df[~df.xyz.isnull()] ある列の値がある値 (仮にabcとする) の行だけ抽出 df = df[df.xyz=='abc'] ある列の値があるリスト (仮にx_list) に含まれる行だけ抽出 df = df[df.xyz.isin(x_list)] 46 / 60
不要な行や列の除外、続き ある列の値がある文字列から始まる行のみ抽出、他 df = df[df.xyz.str.startswith('ABC')] # ABCで始まる df = df[df.xyz.str.endswith('ABC')]
# ABCで終わる df = df[df.xyz.str.contains('ABC')] # ABCを含む (以下、条件部で利用) 文字列のある番号の文字のみ抽出 df.xyz.str[0] # 例、ABC, DEF > A, D 文字列をsplitした場合のある番号のブロックのみ抽出 df.xyz.str.split('-').str[1] # 例、ABC-123, DEF-456 > 123, 456 型変換 df.xyz.astype(int) # 整数に変換 df.xyz.astype(str) # 文字列に変換 47 / 60
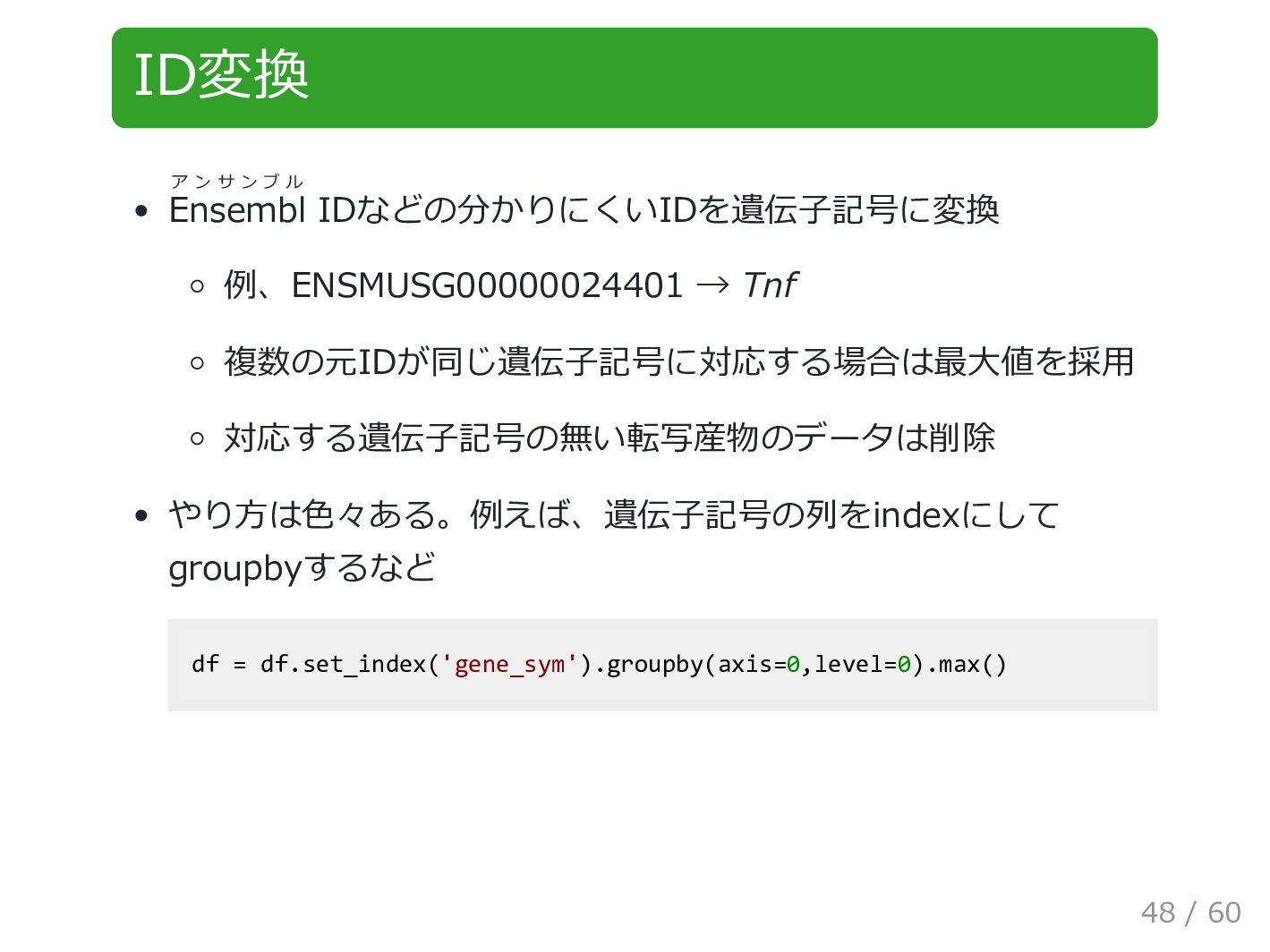
ID変換 Ensembl ア ン サ ン ブ ル IDなどの分かりにくいIDを遺伝子記号に変換 例、ENSMUSG00000024401
→ Tnf 複数の元IDが同じ遺伝子記号に対応する場合は最大値を採用 対応する遺伝子記号の無い転写産物のデータは削除 やり方は色々ある。例えば、遺伝子記号の列をindexにして groupbyするなど df = df.set_index('gene_sym').groupby(axis=0,level=0).max() 48 / 60
列名の変更 必要に応じて自分が分かりやすいように各列の名前を変更する。 手動で元ファイルを変更するのでなく、スクリプトを使う。 ミスを防ぐため、後で結果を再現できるようにするため データが数列程度なら直接書いても可 df.columns = ['ABC','DEF','GHI'] # 3列の列名を更新
長い場合は、for文などで修正名リストを作ってから更新 データの最初だけ見て番号順になっていると過信しない。 正規表現を使わずとも、大抵のことはreplaceとsplitで可能 49 / 60
正規化 マイクロアレイでは分位数正規化 (quantile normalization) など も使われていたが、RNA-seqデータではほぼTPM TPMでは、まず行毎に正規化し、次に列毎に正規化する。 df= df.divide(len_sr, axis=0)
# 各行を遺伝子の長さで割る df = df / df.sum() # 列毎に総和で割る df *= 10**6 # 1Mを掛ける ちなみに、RPKMでは行と列と正規化の順が逆 TPMで箱ひげ図がうまく揃わない場合、私は刈り込みTPMを使っ ている。2行目を以下のように変更 from scipy import stats trim_sum = stats.trim_mean(df, 0.05) * len(df) # 刈り込み総和 df = df / trim_sum # 列毎に刈り込み総和で割る 50 / 60
対数化 0は対数を取れない。 1などを予め足す場合もある。 私は、0は0のままとし、0以外の最小値を0にシフトしている。 df[df==0] = None # 0を一旦Noneに置換 df
= df.apply(np.log2) # 対数化 df -= df.min().min() # 0以外の最小値を引く df = df.fillna(0) # Noneを0に戻す 0以外の場合を式で書くと . ただし、どうしても0と0以外の小さい値の差が不自然に大きく なる問題が生じる。当然、PCAやDEGに悪影響が出る。 結局、対数を取るなら後述する低発現遺伝子の除外が必要 log(x/xmin) 51 / 60
外れ値の除外 PCAとヒートマップを目視で確認し、外れ値のサンプルを除外 PCAでは同じ条件の他の点から遠く離れた位置に来る。 ヒートマップでは縦線が出る。 52 / 60
PCAの使い方 色分け表示するにはseabornのscatterplotが便利 座標データをデータフレーム化し、各サンプルのグループ名を 3列目にして、hueでその列名を指定する。 from sklearn.decomposition import PCA model =
PCA(n_components=2) # PCAオブジェクトを用意 score = model.fit_transform(df.T) # スコア行列の計算 pos_df = pd.DataFrame(score, columns=list('xy')) # データフレーム化 pos_df['group'] = group_list # 各サンプルのグループ名を追加 perc1, perc2 = 100 * model.explained_variance_ratio_ # 寄与率 fig, ax = plt.subplots() # 図を用意 sns.scatterplot(data=pos_df, x='x', y='y', hue='group', ax=ax) # 散布図の描画、色はグループ別 ax.set_xlabel(f'PC1 ({perc1:.1f} %)') # x軸のラベル、寄与率込み ax.set_ylabel(f'PC2 ({perc2:.1f} %)') # y軸のラベル、寄与率込み ax.legend(bbox_to_anchor=(1,1)) # 凡例の位置を枠外に移動 fig.show() # 図の表示 53 / 60
ヒートマップの使い方 RNA-seqデータをヒートマップにする時は遺伝子毎のZスコア化 (標準化) が必須 clustermapの場合 (簡単だが微調整できない) sns.clustermap(df, z_score = 0,
# 各行 (遺伝子) をZスコア化 cmap = 'RdBu_r', # 高発現が赤、低発現が青 vmax = 3, # カラースケールの最大値 vmin = -3, # カラースケールの最小値 row_cluster = True, # 各行をクラスタリングする col_cluster = False, # 各列をクラスタリングしない metric = 'correlation', # 相関係数を類似度とする method = 'average' # 平均連結法を使用 ) 微調整したい場合はsns.heatmapを使う。 54 / 60
ヒートマップの使い方2 sns.heatmapを使う場合 from scipy.cluster.hierarchy import linkage, leaves_list # 行毎にZスコア化 df
= ((df.T - df.T.mean()) / df.T.std()).T # 行方向をクラスタリング、相関類似度と平均連結法を使用 Z = linkage(df, metric='correlation', method='average') # 行方向を並べ替え df = df.iloc[leaves_list(Z)] # 図の描画 fig, ax = plt.subplots() # 図を用意 sns.heatmap(df, cmap='RdBu_r', ax=ax, vmax=3, vmin=-3) # ヒートマップの描画 fig.show() # 図の表示 55 / 60
低発現遺伝子の除外 低発現遺伝子の除外は必要 発現量が小さいほどS/N比が悪い。 0と非ゼロの差が対数化で増幅される。 平均値や中央値が閾値以上の遺伝子のみ抽出 df = df[df.mean(axis=1)>=5] # 平均が5以上の遺伝子を抽出
多臓器データの場合、臓器毎に計算する。 消化酵素など一部の臓器のみで発現する遺伝子がいるため 56 / 60
前処理が妥当かどうかの確認 私は以下の4つで前処理が妥当かどうかを確認している。 箱ひげ図 PCA DEGのヒートマップ DEGのエンリッチメント解析 大事なのは 違和感に気付く こと。 57
/ 60
Outline はじめに 一般的な手法 同期性揺らぎ遺伝子解析 (DNB解析) エネルギー地形解析 RNA-seqデータ前処理 まとめ 58 /
60
まとめ、その1 私の知っている複数のデータ解析手法について紹介した。 一般的な手法として、PCA、階層的クラスタリング、倍率変化に 基づくDEG取得、エンリッチメント解析などを説明した。このあ たりは実験系の方が自分で扱えるようになることが望ましい。 DNB解析が難しい理由として、手法が確立しておらず、選択肢の 組み合わせが膨大で、データ毎に結果が妥当かどうかを判断する 際に高度なドメイン知識が必要なためと説明した。 59 /
60
まとめ、その2 エネルギー地形解析を使うこと自体は簡単だが、解釈が難しいこ とを説明した。逆に言うと、慣れれば武器になる。 RNA-seqデータの前処理をPythonコードと共に説明した。色々 と書いたものの、結局のところ「これをやっておけば大丈夫」と いう保証は無いので、きちんと結果を確認して違和感を見落とさ ないことが何より大事。 60 / 60
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![不要な行や列の除外、続き ある列の値がある文字列から始まる行のみ抽出、他 df = df[df.xyz.str.startswith('ABC')] # ABCで始まる df = df[df.xyz.str.endswith('ABC')]](https://files.speakerdeck.com/presentations/b7e05982f2914a0fae5fb9f2b8a60a6e/slide_46.jpg){kind=link}
{kind=link}
![列名の変更 必要に応じて自分が分かりやすいように各列の名前を変更する。 手動で元ファイルを変更するのでなく、スクリプトを使う。 ミスを防ぐため、後で結果を再現できるようにするため データが数列程度なら直接書いても可 df.columns = ['ABC','DEF','GHI'] # 3列の列名を更新](https://files.speakerdeck.com/presentations/b7e05982f2914a0fae5fb9f2b8a60a6e/slide_48.jpg){kind=link}
{kind=link}
![対数化 0は対数を取れない。 1などを予め足す場合もある。 私は、0は0のままとし、0以外の最小値を0にシフトしている。 df[df==0] = None # 0を一旦Noneに置換 df](https://files.speakerdeck.com/presentations/b7e05982f2914a0fae5fb9f2b8a60a6e/slide_50.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![低発現遺伝子の除外 低発現遺伝子の除外は必要 発現量が小さいほどS/N比が悪い。 0と非ゼロの差が対数化で増幅される。 平均値や中央値が閾値以上の遺伝子のみ抽出 df = df[df.mean(axis=1)>=5] # 平均が5以上の遺伝子を抽出](https://files.speakerdeck.com/presentations/b7e05982f2914a0fae5fb9f2b8a60a6e/slide_55.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}